STORIA DELLA TERAPIA MEDICA IN CARDIOLOGIA
la legge della similarità

Al tempo degli uomini primitivi la terapia si basava su due principi: la legge di similarità e la legge del contagio. La cura veniva tentata attraverso la legge di similarità (similia similibus curantur) mentre la legge del contagio era utilizzata nella formulazione dello stesso agente terapeutico. Le idee magiche dell’uomo primitivo seguivano le due leggi. La storica della medicina Cecilia Mettler (1909-1943) racconta come, nel seguire la legge di similarità, parti delle piante che assomigliavano ad organi o a porzioni corporee venivano considerate esercitare un’influenza su quelli.
Nell’applicare la legge del contagio lei cita come esempio l’uso di pietre lunari nel trattamento di certe forme di pazzia – sulla base della credenza che queste erano causate dalle azioni della luna, e dal momento che anche le pietre lunari si erano formate sotto l’influenza della luna potevano esercitare un effetto curativo.
ASSIRO-BABILONESI
Con l’evoluzione della vita comune e l’affermazione delle pratiche religiose, la farmacoterapia iniziò ad emergere, radicata nel misticismo, nella demonologia, e nella credenza che la malattia fosse causata da una divinità irata.
Nell’era Assiro-Babilonese, la magia e l’acqua erano i principali elementi della materia medica. Oltre all’acqua, i medici dell’era Assiro-Babilonese ricorsero anche all’uso dei prodotti botanici. La farmacopea assiro-babilonese (Shammu) era piuttosto modesta di numero, e le tavolette di quell’epoca elencano poco più di 300 farmaci. Jastrow descrive una prescrizione tipica per quello che sembra essere un processo intra-addominale.
“Se l’interno di un uomo è rigonfio ed infiammato, ed egli è nauseato, allora per la sua vita … cipolla mista a semi di cumino, lasciare che lui beva vino senza mangiare, ed egli si riprenderà. Oppure che prende la scorza verde delle piante, la mischi con grasso di porco, e lasciare che beva con Du-Zab vino puro ed acqua addolcita, ed egli recupererà”.
EGIZI
Il Papiro Ebers menziona più di 700 farmaci che i medici Egizi potevano scegliere. Tra questi c’era l’oppio, l’olio d’oliva, parti di animali, pietre preziose, amuleti, l’assenzio, ed il succo di finocchio fresco. Le erbe, comunque, svolgevano un ruolo predominante nella farmacopea egizia.
“Pharmakon” è il termine greco che indica i farmaci e le sostanze tossiche. Questa è la radice di tutti i nomi ed aggettivi legati ai farmaci. La farmacopea greca consisteva soprattutto di erbe e rappresentò un allargamento della polifarmacia egizia. I derivati delle erbe agivano nella maggior parte dei casi come agenti mediatori dal momento che i loro effetti terapeutici erano intimamente collegati con le credenze religiose.
antichi greci
Il politeismo pagano degli Antichi Greci creò ulteriore confusione con invocazioni rivolte a diversi dei come Apollo, Zeus ed il centauro Chirone. L’avvento degli Asclepiadi ed il crescente numero di seguaci di Ippocrate determinò una graduale semplificazione di questa pesante polifarmacia. Essi utilizzarono soprattutto delle pratiche misure correttive, ponendo enfasi sulla dieta, il cambio del clima, agenti purgativi e naturali senza ricorrere alle pietre preziose, agli amuleti ed altri agenti per scacciare i demoni. Il miele nell’aceto era ritenuto avere proprietà espettoranti e diuretiche. Ippocrate scrisse che l’ossimele (il miele nell’aceto):
“promuove l’espettorazione e libera il respiro … Esso promuove anche scariche flatulenti dell’intestino, ed è diuretico, … e diminuisce la forza e rende fredde le estremità. Questo è il solo cattivo effetto degno di menzione che io so derivare dall’ossimele”.
Durante tale era, Teofrasto (371 ac – 287 ac) fu il principale esponente dei rimedi botanici. Egli era un pupillo di Aristotele, e sfruttò appieno l’intero giardino botanico di Aristotele concessogli dallo stesso maestro. Teofrasto descrisse circa 500 farmaci derivati dalle piante. Egli affermava che la robbia, che chiamò “ereuthedanon”, aveva proprietà analgesiche e diuretiche.
Gli Alessandrini
Il Periodo Alessandrino è noto per la sua enfasi verso l’uso dei veleni ed i loro antidoti. Anche alcuni re furono coinvolti in questo aspetto atroce della materia medica.
Il più noto fu Mitridate, il sesto re del Ponto. Secondo la leggenda egli era ben istruito sui veleni ed aveva anche “concotto” un antidoto universale. Questo era fatto con foglie di ruta, noce, sale e fico. Per essere efficace doveva essere preso giornalmente. Mitridate occupò un ruolo importante in una parte della storia romana. Questo farmaco fu modificato da Andromaco il Vecchio, archiatra di Nerone, con l’aggiunta di parti di scilla, carne di vipera ed oppio. Questa “concozione” fu descritta a Nerone come prima linea di difesa contro gli avvelenamenti mortali. Andromaco la raccomandò anche come agente efficace nell’edema generalizzato ed in altre condizioni.
L’intera concozione modificata divenne nota con il nome di teriaca e, con l’eponimo “theriaca andromachi” o anche triaca di Venezia, e rimase un importante agente terapeutico fino alla metà del 1700 dc.
Gli antichi romani
In Epoca Romana, Il V libro del De Medicina di A. C. Celso è dedicato interamente alla terapia. Dal punto di vista dei proprietari terrieri e di schiavi, questa era probabilmente la parte più utile dell’opus di Celso. Il libro contiene 28 capitoli e descrive una considerevole varietà di agenti terapeutici. Le fonti di provenienza sono varie. Riguardo al trattamento delle malattie cardiache viene fatta una scarsa menzione; probabilmente perché le varie manifestazioni delle disfunzioni cardiache erano poco conosciute a quell’epoca.
Dioscoride Pedanio (40 dc circa – 90 dc circa), nativo di Anazarba, in Cilicia, scrisse un trattato che rappresenta la quintessenza della farmacologia del periodo. Dioscoride è famoso per la sua opera in 5 libri, De Materia Medica, un erbario scritto in lingua greca che ebbe una profonda influenza nella storia della medicina. Rimase infatti in uso, con traduzioni e commenti, almeno fino al XVII secolo. Oltre che in area greca e romana, quest’autore fu conosciuto anche in Oriente e sono rimasti svariati manoscritti di traduzioni arabe e indiane.
L’intera farmacopea di Galeno si basava principalmente sull’ipotesi umorale dell’era ippocratica che allora era associata alla teoria dei quattro elementi. Egli scrisse sull’argomento circa 30 libri. Galeno aveva una scarsa fiducia verso gli altri al punto da possedere un proprio deposito in cui raccoglieva una vasta quantità di farmaci ed erbe. Nei suoi viaggi egli cercava sempre di aggiungere nuovi argomenti alla sua materia medica. Galeno preparava la sua triaca con più di 100 ingredienti, e tra questi vi era la squilla. Galenici erano la maggior parte dei medicinali che venivano utilizzati fino al XVIII secolo; un altro esempio del profondo rapporto di Galeno con la pratica medica.
I Bizantini
La materia medica dell’era bizantina fu caratterizzata dall’adesione alla fede superstiziosa verso le procedure magiche in associazione con le prescrizioni galeniche.
Paolo di Egina (625 circa – 690 circa) spicca in questa epoca con il suo testo medico consistente di 7 libri, l’ultimo dei quali dedicato alla materia medica. Egli raccomanda di bere sangue di capra con latte per il trattamento dell’edema.
Gli Arabi
I Bizantini trasmisero la loro conoscenza medica agli Arabi e Dioscoride fu praticamente la fonte della loro intera tradizione medica. Essi introdussero l’ambra grigia per il trattamento dei crampi, delle malattie cardiache e i disordini cerebrali, ed il macis per le malattie cardiache e l’indigestione.
La sezione dedicata alla materia medica dell testo di Avicenna (o Ibn-Sina), Al-Quanum-fi al-Tibb (anche noto come il Canone), elenca 760 farmaci, raccolti da fonti bizantine e persiane di agenti hindu. Il Canone di Avicenna esercitò un’importante influenza sulla medicina occidentale fino al XVII secolo. Occasionalmente esso viene ancora ripreso nei paesi mussulmani. Le allusioni ai rimedi per la malattia cardiaca sono ripetizioni di quelle che si trovano in Dioscoride. La sezione del libro di Avicenna sui farmaci per il cuore ne elenca in totale 63.
Alto Medioevo
Durante questo periodo del Medioevo, il mondo occidentale continuò a basare la sua farmacopea sulle radici greco-romane e Dioscoride rimase la principale autorità. Le variazioni cristiane ai culti pagani introdussero nuove componenti magiche alle pratiche di superstizione. Gli dei pagani del culto di Esculapio furono sostituiti dai santi cristiani. Sebbene i manoscritti medievali non introdussero nuovi importanti concetti medici, essi ebbero la funzione di codificare le informazioni da una generazione all’altra. In particolare, i monaci furono i più attivi nel copiare e tradurre i manoscritti. Più di ogni altro, in questo campo, è degno di nota Aurelio Cassiodoro (480-575). Mentre era nel monastero benedettino di Monte Cassino, egli scrisse i seguenti passi di farmacologia:
“Impara a conoscere le proprietà delle erbe e come mescolare i farmaci, ma riponi le tue speranze nel Signore, che preserva la vita in modo continuo. Se la lingua dei Greci ti è sconosciuta, hai il libro delle erbe di Dioscoride, che ha descritto e mostrato le erbe in maniera molto accurata. Dopo leggi la traduzione latina di Ippocrate e Galeno, in particolar modo il libro di terapia che quest’ultimo ha dedicato al filosofo Glaucone, ed il lavoro di un anonimo autore che, come sembra, è compilato da diversi scrittori. Studia inoltre la medicina di Aurelius [Aurelianus] Caelius, il libro ippocratico sulle erbe e sui metodi curativi, ed anche una varietà di trattati sull’arte di guarire che ho messo insieme nella mia libreria e ti ho tramandato.”
scuola salernitana
La Scuola Salernitana esercitò la sua influenza sulla farmacologia soprattutto durante l’undicesimo ed il dodicesimo secolo. In questa scuola emersero due importanti autori: Nicolaus Salernitanus e Matteo Platearius (Salerno, XI secolo – XII secolo). Anche in questo ambiente nessuna nuova scoperta venne fatta, ed il loro lavoro fu una reiterazione della terapia tradizionale di Dioscoride.
letteratura inglese
Il primo libro pubblicato in inglese sulle erbe fu stampato da Richard Bankes, di Londra. Questo lavoro, conosciuto a con come “Banckest Herbal” (1525), contiene i caratteri dei primi libri medici e manoscritti di ricette anglosassoni.
Rinascimento
Con l’avvento del Rinascimento, l’alchimia iniziò a diventare parte integrante della farmacopea. Ma ciò non fu semplice. Questa dovette combattere contro la ripresa d’interesse verso le preparazioni botaniche a causa della scoperta ed esportazione di molti nuovi prodotti del Nuovo Mondo.
Nicolas Monardes (1493 – 10ottobre 1588) fu un promotore attivo dell’introduzione di nuove erbe e piante con proprietà medicinali provenienti dall’India e dall’America. Anche il tabacco importato dall’America fu reclamizzato come avente marcate proprietà curative. Jean Nicot (1530 – 10maggio 1604), l’ambasciatore francese in Portogallo, importò e coltivò il tabacco nel suo paese nativo. Non a caso la nicotina prende ora il suo nome. Mentre l’Europa veniva inondata di prodotti botanici provenienti dal Nuovo Mondo, gli alchimisti continuarono a lavorare assiduamente nella loro ricerca della pietra filosofale e l’elisir di lunga vita. Paracelso catalizzò l’interesse verso i rimedi chimici. Egli introdusse il “Lilium Paracelsi” come un costituente di un elisir teriacale. Questo conteneva stagno, rame e una lega di ferro e antimonio. Riguardo ai presunti effetti benefici e tossici dell’antimonio derivò una controversia che si prolungò nel tempo.
Il seicento
Theodore Turquet de Mayerne (28settembre 1573 – 22marzo 1655), di Ginevra, introdusse, durante la prima parte del XVII secolo un certo ordine nella preparazione e diffusione dei farmaci in Inghilterra. Egli ebbe il potere di fare ciò grazie alla sua posizione di medico reale alla corte di Giacomo I e Carlo I d’Inghilterra. Il suo passo iniziale nel districarsi dalla confusione fu quello di separare la corporazione dei droghieri da quella dei farmacisti. Contemporaneamente si dovettero stabilire i criteri di preparazione dei vari medicamenti. La Pharmacopeia Londinensis, un libro di riferimento che de Mayerne sponsorizzò, si adattò perfettamente a questo scopo. Il libro non era nient’altro che una copia della VI edizione della AugsburgPharmacopeia, e sebbene non avesse alcun merito di originalità, acquisì una reale significatività dopo alcune revisioni ed edizioni.
IL SETTECENTO
Il XVIII secolo vide una maggiore cooperazione tra chimici ed erboristi. Ciò non è sorprendente dal momento che molti progressi fatti dai chimici furono possibili grazie a campioni botanici grezzi che erano la fonte dei loro agenti purificati. Comunque, si dovettero attendere parecchi anni prima che i chimici riuscissero a sintetizzare agenti chimici puri senza il bisogno di erbe o piante.
L’OTTOCENTO
Se non ad altro, questa analisi storica dovrebbe essere servita ad enfatizzare il fatto che, fino al XIX secolo, la terapia medica delle malattie cardiache era virtualmente inesistente. Erano disponibili pochi agenti ed in composizioni rozze, poco efficaci e ricche di effetti indesiderati. I salassi erano una tecnica terapeutica popolare e tenuti in considerazione principalmente grazie agli insegnamenti di Jean-Nicolas Corvisart des Marets (15febbraio 1755 – 18settembre 1821), Jean-Baptiste Bouillaud (16settembre 1796 – 29ottobre 1881) ed altri luminari del tempo.
Catartici idragoghi contenenti sale vennero usati nel trattamento dell’edema generalizzato. La squilla, sopravvissuta dai tempi degli antichi Egizi ed ancora ubiquitaria come ingrediente della triaca, veniva usata in modo assurdo. Si utilizzavano anche le foglie della digitale ma senza alcuna conoscenza delle sue azioni farmacologiche e senza alcuna giusta ragione nonostante la monumentale monografia di William Withering (17marzo 1741 – 6ottobre 1799) sull’argomento (“An Account of the Foxglove, and Some of its Medical Uses“).
Un quadro analitico della materia medica nel campo della patologia cardiaca si può avere prendendo come riferimento il testo “Diseases of the Heart” di James Hope, pubblicato nel 1839. Il mercurio competeva con l’antimonio come ancora di salvataggio del trattamento. Questo era disponibile in forma di “pillola azzurra” o calomelano.
La iosciamina e la digitale venivano somministrati per gli attacchi nervosi e l’endocardite acuta. Il termine idropisia indicava in maniera generale la ritenzione dei fluidi a prescindere dalle cause, e veniva trattata con una serie di agenti che avevano proprietà diuretiche come la squilla, il ginepro, i tartrati, gli acetati, il decotto di ginestra, gli spiriti di etere nitroso, il nitrato di potassio ed i cantaridi.
La digitale ed i salassi
La foglia di digitale veniva usata frequentemente nell’idropisia secondo la credenza che fosse un agente diuretico piuttosto che un farmaco cardiotonico. La digitale veniva usata anche per le palpitazioni ma in base all’errata nozione che questo disordine fosse dovuto ad uno stato nervoso piuttosto che ad una sottostante aritmia. Nessun farmaco specifico veniva usato per l’ipertrofia cardiaca o la dilatazione cardiaca, essendo il secondo un sinonimo di scompenso cardiaco.
I salassi erano ancora “di rigore” per l’ipertrofia cardiaca, perlomeno durante la prima metà del XIX secolo. Involontariamente, tale pratica, riducendo il volume sanguigno, poteva avere qualche effetto in quelle forme di ipertrofia ventricolare che erano una conseguenza dell’ipertensione.
Gli obiettivi terapeutici nell’aneurisma aortico erano quelli di ridurre la frequenza e la contrattilità cardiaca. La digitale veniva erroneamente somministrata come agente inotropo negativo simultaneamente ai diuretici ed i purganti.
Ippolito Francesco Albertini (Crevalcore, 26ottobre 1662 – Bologna, 26marzo 1738) e Antonio Maria Valsalva (Imola, 17gennaio 1666 – Bologna, 2febbraio 1723) sostenevano la pratica dei salassi vigorosi. Hope aderì al loro consiglio ma lo temperò riducendo la quantità di sangue estratto e la frequenza della sua rimozione. Le sanguisughe venivano usate in aggiunta alla flebotomia.
angina pectoris
Il XIX secolo vide l’evoluzione del trattamento antianginoso progredire in vari stadi e con vari farmaci che furono trattati su basi empiriche o su errati concetti fisiopatologici. La nozione prevalente prima di James Bryan Herrick (11agosto 1861 – 7marzo 1954) e William Heberden era che la malattia cardiaca raramente presentava dei sintomi. Più spesso veniva ritenuto colpevole lo stomaco, e la terapia veniva diretta verso il sollievo dall’acidità, dai gas o dai contenuti gastrici indigesti. L’acidità veniva combattuta con soda o preparati a base di gesso. Venivano somministrati anche agenti antispasmodici in base alla credenza che il tratto gastro-intestinale fosse la sede del problema. Questi farmaci venivano somministrati in forma di impiastri a base di belladonna. Venivano usati anche estratti o tinture di oppio soprattutto per l’azione sedativa.
Lo stesso Laénnec credeva che l’angina pectoris avesse un’origine neurologica. Egli usava due piastre d’acciaio fortemente magnetizzate, una sul precordio e l’altra sull’area scapolare di sinistra. Questa forma di terapia sembrava avere effetti positivi solo nelle mani di Laénnec.
I NITRATI
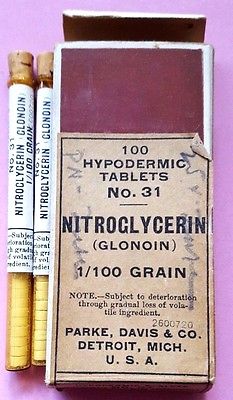 L’uso di un gruppo nitrogeno di origine chimica per il trattamento dell’angina pectoris iniziò con la scoperta della nitroglicerina. Dal momento che la storia dell’applicazione medica della nitroglicerina è intimamente collegata con quella dell’omeopatia, è appropriato, in questo momento, un breve discorso su questa branca (?) della terapia medica. Samuel Hahnemann, di Meissen (10aprile 1755 – 2luglio 1843), fu colui che introdusse l’omeopatia. La scoperta del chimico tedesco Christian Friedrich Schönbein (18ottobre 1799 – 29agosto 1868) riguardante la potenza esplosiva del “gun cotton” (fulmicotone o nitrocellulosa) fu lo stimolo per la ricerca di un altro esplosivo con gruppo nitrogeno. Questa ricerca portò alla sintesi della nitroglicerina nel 1846 per opera di Ascanio Sobrero (12ottobre 1812 – 26maggio 1888), un chimico italiano.
L’uso di un gruppo nitrogeno di origine chimica per il trattamento dell’angina pectoris iniziò con la scoperta della nitroglicerina. Dal momento che la storia dell’applicazione medica della nitroglicerina è intimamente collegata con quella dell’omeopatia, è appropriato, in questo momento, un breve discorso su questa branca (?) della terapia medica. Samuel Hahnemann, di Meissen (10aprile 1755 – 2luglio 1843), fu colui che introdusse l’omeopatia. La scoperta del chimico tedesco Christian Friedrich Schönbein (18ottobre 1799 – 29agosto 1868) riguardante la potenza esplosiva del “gun cotton” (fulmicotone o nitrocellulosa) fu lo stimolo per la ricerca di un altro esplosivo con gruppo nitrogeno. Questa ricerca portò alla sintesi della nitroglicerina nel 1846 per opera di Ascanio Sobrero (12ottobre 1812 – 26maggio 1888), un chimico italiano.
Constantin Hering (1gennaio 1800 – 23luglio 1880), un medico omeopata, nato in Germania e residente a Philadelphia, fu il primo tra gli omeopati ad intravedere le possibilità terapeutiche della nitroglicerina. Egli ed i suo gruppo di colleghi omeopati condussero le prime osservazioni su se stessi. Tale approccio non era nuovo tra gli omeopati. Lo stesso Hahnemann praticava routinariamente l’auto-sperimentazione quando valutava il valore terapeutico dei vari agenti. Hering utilizzò per la nitroglicerina l’acronimo “glonoina”. Dopo un anno di provini egli concluse che:
“Questa nuova sostanza ha causato mal di testa in tutti coloro che l’anno usata: perciò … essa curerà i mal di testa ed gli altri malanni che sono simili nei sintomi a quelli prodotti da questa in condizioni di salute.”
Egli osservò anche:
“Il tipo di mal di testa, avendo un altissimo grado di pulsatilità, mi ha condotto ad esaminare il polso, ed in tutti i casi il polso era alterato. Controllando il polso ho scoperto che la glonoina, anche i quantità molto piccole, esercitava una maggiore influenza sull’attività cardiaca rispetto ad ogni altro rimedio usato prima d’ora. Il polso si accelerava in tutte le prove, anche immediatamente, non appena i globuli venivano sciolti sulla lingua.”
Il paziente che provava l’effetto clinico del farmaco veniva chiamato “prover”. La glonoina di Hering era una soluzione alcolica di nitroglicerina imbevuta in una pallottolina di zucchero. La glonoina divenne un rimedio omeopatico diffuso per molte condizioni ma mai per l’angina pectoris.
Alfred Field, un britannico, fu il primo tra i medici allopati a studiare in profondità gli effetti clinici della nitroglicerina. Egli pensava che esso fosse un antispastico gastrointestinale e lo usò per la prima volta su una donna di 69 anni che aveva frequenti episodi di “intenso dolore epigastrico, che si estendeva alla parte superiore del torace ed lato interno del braccio sinistro”. Field pubblicò il suo articolo nel 1858. Questo giunse all’attenzione di molti altri medici britannici che aggiunsero le proprie osservazioni a quelle di Field.
Nel 1864, il medico tedesco J. Albers pubblicò la prima vera valutazione scientifica della nitroglicerina. Questo era uno studio fisiologico e terapeutico che enfatizzava i suoi effetti sul cuore e sulla circolazione.
Dal 1879, William Murrell (1853-1912) fu il primo medico allopata che sostenne con forza l’uso della nitroglicerina nell’angina pectoris. La sua analisi sull’argomento comparve su The Lancet. Furono studiati trentacinque pazienti, e tutti descrissero la cefalea, tachicardia e palpitazioni così come era accaduto per sé stesso. Murrell, in successivi studi, documentò le modificazioni del polso con lo sfigmometro di recente invenzione. Egli annotò una similarità d’azione tra la nitroglicerina ed il nitrito d’amile, che lo condusse a commentare:
“Considerando l’azione fisiologica del farmaco e, soprattutto, la somiglianza tra la sua azione generale con quella del nitrito d’amile, ritengo di poter dire che questa sia un prova a favore del trattamento contro l’angina pectoris, e perciò ho già iniziato ad usare tale farmaco in questa situazione”.
Già un anno dopo l’articolo di Murrel, nel 1880, un chimico britannico, William Martindale preparava la nitroglicerina in compresse alla dose di 1/100 di un grano, il sistema farmaceutico posologico in uso allora e corrispondente a circa di 0,64 milligrammi. La compressa di Martindale si dissolveva rapidamente in bocca con un rapido assorbimento ed un immediato sollievo dei sintomi. Egli ne garantì la sicurezza ed affermò che era stabile, non volatile e non poteva essere detonata se assunta in questa maniera. Parke-Davis fu la prima casa farmaceutica americana a preparare e commerciare la nitroglicerina in forma di compresse.
Nel 1883, Mathew Hay pubblicò un articolo in cui descriveva i suoi esperimenti sulla farmacologia della nitroglicerina. Egli descrisse la natura chimica e l’azione fisiologica di tale agente.
il novecento
Le tecniche sperimentali del XX secolo, soprattutto l’angiografia, hanno provato le proprietà vasodilatatrici dei nitrati e della nitroglicerina così come dei loro effetti emodinamici sul preload e l’afterload.
I nitrati a lunga durata d’azione comparvero negli anni quaranta del novecento. In questo periodo molte importanti case farmaceutiche rivolsero la loro attenzione allo sviluppo di farmaci in grado di sostenere la vasodilatazione delle arterie coronariche.
Il XX secolo vide la diffusione di una valanga di agenti farmaceutici nella gestione dei disturbi cardiaci.
LA DIGITALE
All’età di 35 anni, nel 1775, William Withering (1741-1799) pubblicò una classificazione degli elementi botanici della Gran Bretagna. Questa s’intitolava “A Botanical Arrangement of All The Vegetables Naturally Growing in Great Britain”. In questo libro Withering descrisse per la prima volta le potenzialità terapeutiche della digitalis purpurea. Per quanto riguarda la cardiologia, il capolavoro di Withering fu la sua monografia intitolata “An Account of the Foxglove And Some of its Medical Uses: with Pratical Remarks on Dropsy, And Other Diseases”. Quest’opera fu pubblicata nel 1785. Withering descrisse in dettaglio la sua esperienza con il farmaco su una serie di 163 pazienti nell’arco di nove anni. Nel suo trattato furono svelati molti importanti elementi. Egli scoprì che l’ingrediente attivo della digitalis purpurea era la digitale; l’uso di una preparazione standardizzata per un’appropriata valutazione della relazione dose-risposta e descrisse gli effetti del farmaco con le sue manifestazioni tossiche. Withering usò il farmaco principalmente nell’idropisia attribuendo in maniera errata gli effetti favorevoli all’azione diuretica piuttosto che a quella cardiotonica. Solo una successiva ricerca eseguita da altri identificò la digitale come glicoside cardiaco.
digitalis purpurea
Probabilmente la pianta contenente glicosidi cardiaci conosciuta da più lungo tempo è la scilla. Menzioni di questa pianta si possono trovare nel papiro di Ebers e nel “De Universa Medicina”, l’enciclopedica materia medica di Dioscoride.
Né la sua reale composizione chimica né la sua azione sul cuore erano note in quei tempi. Questa veniva usata come ingrediente nella preparazione dei farmaci teriacali. L’uso della digitale risale ad un epoca successiva.
Il nome deriva dal termine latino digitalis purpurea che il botanico tedesco Leonhart Fuchs (17gennaio 1501 – 10maggio 1566) descrisse per la prima volta nel 1542. Nel suo “De Historia Stirpium Commentarii”, Fuchs ascrisse alla pianta sia la proprietà purgativa che emetica. Egli pensava anche che fosse utile nell’idropisia e la raccomandava in forma di decotto o infusione.
Quando Withering introdusse la digitalis purpurea come rimedio di successo nel trattamento dell’idropisia, egli trattò il problema della posologia in modo perspicace, riducendo il dosaggio in maniera decrescente in modo tale da controllare la frequenza delle reazioni tossiche. Egli apprese tale strategia con l’esperienza dal momento che all’inizio della sua pratica gli effetti tossici del farmaco erano eccessivi. Alla fine giunse alla dose di 65 mg (1 grano) due volte al giorno misto con oppio per controllare la nausea ed il vomito.
Quattro anni dopo la pubblicazione della monografia di Withering, la digitale venne inserita nella Infirmary Pharmacopeia di Edimburgo. Jonathan Stokes (c. 1755 – 30aprile 1831) fu colui che allertò i medici di Edimburgo verso questo medicamento.
Nel 1799, John Ferriar descrisse gli effetti cardiaci nel suo “An essay on the medical properties of digitalis purpurea or foxglove”. Egli scrisse:
“gli estratti delle foglie ci forniscono uno strumento per regolare il polso secondo il nostro desiderio o supportarlo nella maniera che riteniamo più appropriata”.
L’effetto inotropo positivo della digitale fu riconosciuto da un altro contemporaneo, Thoma Beddoes.
Jean-Baptiste Bouillaud, nel 1835, descrisse dei casi in cui la digitale controllava un rapido polso intermittente irregolare. Fu a causa di questo effetto tranquillizzante sul battito cardiaco che Bouillaud chiamò la digitale “l’oppio del cuore”. Gli esperimenti di Frank nel 1895 indicarono che la digitale incrementava la portata cardiaca solo nel cuore compromesso e non aveva effetto sulla gittata del cuore normale di rana.
Gold e Cattell nel 1940 dimostrarono senza ombra di dubbio che la digitale aveva un’azione inotropa positiva sul muscolo cardiaco. Essi usarono il muscolo papillare del gatto per descrivere il meccanismo con cui la digitale aboliva l’insufficienza cardiaca.
Mason ed Eugene Braunwald (nato il 15 agosto 1929), oltre che Smith ed Haber, riassunsero rispettivamente nel 1968 e nel 1973 le opinioni correnti sulla digitale. Secondo queste tesi la digitale ha un effetto inotropo positivo sia sui pazienti con scompenso cardiaco che in quelli con normale ritmo sinusale. Questa è la sua azione primaria. Il suo effetto cronotropo è esercitato attraverso il nervo vago e attraverso un’azione diretta depressiva sulla conduzione atrioventricolare. Comunque, entrambe i suoi effetti inotropo e cronotropo possono essere modificati dall’azione della digitale sui riflessi autonomici, sui vasi sanguigni e sulla conduzione.
LE CATECOLAMINE
Esistono molte sostanze derivate dalla amine che hanno anche proprietà inotrope positive come la digitale, ma differiscono da questa in quanto causano aumento della frequenza cardiaca. Tali sostanze sono classificate come catecolamine ed includono l’epinefrina e la norepinefrina oltre che la dopamina, la dobutamina ed il milrinone.
Tra questi farmaci il più vecchio è l’epinefrina, nota anche come adrenalina. Nel 1894 ed il 1895, George Oliver e E. Schafer estrassero dalla ghiandola surrenale una sostanza che aveva delle proprietà farmacologiche molto interessanti. Essi scoprirono che la somministrazione per via intravenosa nel coniglio di una grande quantità di tale estratto surrenalico ne determinava la morte. D’altra parte, la somministrazione di una piccola quantità causava un immediato e marcato aumento della pressione arteriosa. Esperimenti successivi indicarono che l’aumento della pressione arteriosa era dovuta ad incremento della resistenza periferica attraverso una generalizzata costrizione arteriolare. Essi non conoscevano il principio attivo ma erano certi che questo provenisse dalla porzione midollare della ghiandola surrenale.
La loro scoperta suscitò un grande interesse tra i fisiologi, e quelli che erano più predisposti verso la chimica cercarono di isolare il principio attivo. Dopo due anni, nel 1897, John Jacob Abel (19maggio 1857 – 26maggio 1938), che fu il primo professore di farmacologia dell’emisfero occidentale, presentò un articolo all’Association of American Physicians in cui descrisse il suo metodo per isolare il principio attivo che chiamò epinefrina.
Il 5 novembre 1900, Jokichi Takamine (3novembre 1854 – 22luglio 1922) richiese il brevetto americano per un estratto di ghiandola surrenale che egli chiamò “adrenalina”. Sei mesi dopo gli venne assegnato il diritto di usare la parola “adrenalina” come marchio di fabbrica. In seguito Takamine assegnò la licenza del prodotto alla Parke-Davis & Co.
Takamine fece fortuna mentre Abel rimase un accademico che cercava il modo di purificare la sua epinefrina e che ingaggiò diverse controversie con alcuni ricercatori tra cui, ovviamente, lo stesso Takamine. Abel asserì in modo persistente che l’adrenalina di Takamine non era pura, ed in ciò egli aveva ragione poiché studi successivi dimostrarono che essa conteneva una certa quantità di norepinefrina. La controversia scemò allorchè Abel rivolse la sua attenzione ad altri progetti.
Nel 1905, l’articolo di Eliot sull’azione dell’adrenalina comparve sul Journal of Physiology. Egli sottolineava gli effetti inotropi e cronotropo positivi dell’adrenalina; attributi eccellenti ma deturpati dalla sua propensione ad indurre aritmie. Fu in particolare questa reazione avversa che determinò una nuova ricerca di nuovi agenti inotropi nella famiglia delle catecolamine.
Goldberg descrisse nel 1972 l’azione cardiovascolare e renale della dopamina con potenziali applicazioni cliniche.
R. R. Tuttle, nel 1975, descrisse la capacità di un’altra catecolamine, la dobutamina, di incrementare in maniera selettiva la contrattilità cardiaca.
I CALCIO-ANTAGONISTI
La storia dei Ca-antagonisti è collegata in modo particolare agli sforzi di un uomo, il farmacologo tedesco Albrecht Fleckenstein (3marzo 1917 – 4aprile 1992). L’identificazione e lo sviluppo iniziarono e continuarono nel suo laboratorio all’Istituto Fisiologico dell’Università di Friburgo. In un articolo sulla storia dei calcio antagonisti egli ne divise l’evoluzione in tre periodi.
- Il primo periodo è costituito dagli eventi che hanno determinato la nascita del concetto di calcio antagonismo.
- Gli studi sul meccanismo ed i siti d’azione costituiscono il secondo periodo,
- mentre il terzo riguarda l’accettazione dei principi e lo sviluppo dei congeneri.
Due compagnie farmaceutiche (Knoll e Hoechst) furono coinvolte in modo particolare dallo sforzo di Fleckenstein. Riguardo a ciò Fleckenstein affermò:
“La scoperta del calcio-antagonismo avvenne in modo casuale. Ciò avvenne nel novembre del 1963, allorchè mi fu richiesto da due case farmaceutiche tedesche (Knoll e Hoechst) di dare un’occhiata su due vasodilatatori coronarici di nuova sintesi con inspiegati effetti cardiodepressivi. Uno di questi composti era la prenilamina. L’altro composto non aveva ancora un nome, ma fu chiamato in seguito Isoptin, Iproveratril, o con il nome generico di verapamil. Con nostra sorpresa, divenne subito chiaro che entrambi i composti mimetizzavano gli effetti cardiaci del semplice ritiro di Calcio. Come ho riportato nel 1964, entrambe le sostanze inibivano l’accoppiamento cardiaco eccitazione-contrazione in quanto diminuivano la forza contrattile senza aumentare il potenziale d’azione (Fleckenstein, 1964). Inoltre, entrambe le sostanze diminuivano l’utilizzazione di fosfato ad alta energia Calcio dipendente nei ventricoli battenti e riducevano il consumo di ossigeno in maniera parallela alla depressione dell’attività contrattile. Comunque, tutti questi effetti inibitori dei due farmaci potevano essere prontamente neutralizzati con l’aiuto di Calcio addizionale, catecolamine beta-adrenergiche, glicosidi cardiaci e, per così dire, misure che incrementavano o ripristinavano l’apporto di Calcio al sistema contrattile. Tali osservazioni ci condussero a supporre che l’azione comune del verapamil e della prenilamina potesse consistere nell’interferire con la funzione mediatrice del Calcio nell’accoppiamento eccitazione-contrazione del muscolo cardiaco. Nei fatti, l’ipotesi di un’attività di beta-blocco da parte dei due farmaci, come inizialmente proposta dalle due compagnie, fu definitivamente invalidata nel 1967. Dunque, in questo modo nacque il principio farmacodinamico del Calcio antagonismo.”
Nel 1968, il capo chimico della Knoll company, il dr Ferdinand Dengel, presentò a Fleckenstein un metossi derivato del verapamil che era stato chiamato D600. Il termine D600 nasceva dal fatto che questo era il seicentesimo composto sintetizzato da Dengel. In seguito venne chiamato gallopamil. Un anno dopo Fleckenstein ricevette la visita di un altro amico. Costui era il professor Kroneberg, un farmacologo della Bayer. Egli aveva con sé due flaconi recanti il contrassegno “Bay a 1040” e “Bay a 7168”, entrambi vasodilatatori coronarici ma con una marcata azione inotropa negativa. Kroneberg cercava un chiarimento a questa doppia azione. Fleckenstein riuscì a dimostrare che questi due agenti avevano un meccanismo d’azione in comune con il verapamil, la prenilamina ed il D600. Egli decise di chiamare questa intera famiglia di nuovi vasodilatatori “calcio-antagonisti”. Il vero nome dei farmaci della Bayer fu gelosamente tenuto segreto e, solo dopo la pubblicazione dei report di Fleckenstein, divenne noto che il nome della Bay 1040 era nifedipina mentre quello della Bay a 7168 era nilupidina.
Bender, nel 1966, fu il primo a dimostrare gli effetti antiaritmici del verapamil. Shamrot confermò tale dato nel 1972, e negli anni successivi, il verapamil mostrò la sua efficacia nel trattamento delle tachicardie sopraventricolari. Non tutti i Calcio antagonisti hanno questo effetto terapeutico. Studi elettrofisiologici intrapresi nel 1970 mostrarono che alcuni Calcio antagonisti già sul mercato avevano un effetto depressivo sull’automaticità del nodo SA e sulla conduzione atrioventricolare. Contributi alla comprensione di questa modalità d’azione arrivarono da Zipes e Fischer nel 1974 e, sempre nel 1974, da Wit e Cranefield.
Dal 1970, anno d’inizio del terzo periodo di Fleckenstein, c’è stato un incredibile aumento nell’uso degli agenti bloccanti i canali del calcio. Tali farmaci hanno dimostrato la loro capacità nel contrastare gli episodi anginosi, diminuire la pressione sanguigna in virtù delle loro proprietà di dilatazione arteriosa, proteggere la degenerazione del miocardio dal sovraccarico di Ca, e controllare certe aritmie.
I BETA-BLOCCANTI
I beta-bloccanti furono introdotti in cardiologia negli anni sessanta del novecento. Da allora essi hanno assunto un ruolo importante nella gestione farmacologica dell’ipertensione, malattia cardiaca ischemica e certe aritmie.
Raymond P. Ahlquist postulò per primo l’ipotesi che l’attività catecolaminica fosse mediata da recettori alfa e beta adrenergici. Ciò accadeva nel 1948. Le azioni apparentemente contraddittorie delle varie catecolamine potevano essere spiegate solo con questa ipotesi. Per esempio, si scoprì che l’epinefrina poteva eccitare ma anche inibire il muscolo liscio. Ahlquist teorizzò che se l’epinefrina si attaccava a certi recettori localizzati nel muscolo liscio bersaglio che egli chiamò alfa, avveniva la costrizione. D’altra parte, la stimolazione dei recettori beta determinava il rilassamento.
Nel 1958, Powell e Slater sintetizzarono un’agente con capacità beta-bloccante. Questo era il dicloroisoprotenerolo. Sfortunatamente, tale composto era anche un agonista parziale che si mostrò poco affidabile per l’uso clinico visti i suoi effetti collaterali. Comunque, la sintesi di tale farmaco costituì la base per altri studiosi che cercavano un composto senza alcuna azione di agonismo catecolaminico.
Il primo di tale prodotti fu introdotto da James Whyte Black (14giugno 1924 – 22marzo 2010) ed associati alla fine degli anni cinquanta del novecento. Il farmaco fu chiamato pronetalolo. Ma anche questo non poté essere usato clinicamente perché produceva tumori nei topi.
Nel 1962 Black e Stephenson descrissero le loro osservazioni con il propanololo. Essi scoprirono che il propanololo era un antagonista beta-adrenergico competitivo senza alcuna attività agonistica e nessun significativo effetto avverso. Questo farmaco fu messo in commercio con il marchio Inderal, ed in poco tempo divenne il prototipo di numerosi congeneri.
DIURETICI
Lo sviluppo dei veri diuretici è un fenomeno del XX secolo e rappresenta un trionfo della chimica sintetica. Comunque, il progresso della chimica richiese la comprensione dei meccanismi riguardanti la filtrazione glomerulare e il riassorbimento di sale ed acqua. Tale conoscenza fu resa possibile dal farmacologo statunitense Alfred Newton Richards (22marzo 1876 – 24marzo 1966) negli anni trenta del novecento. Egli sottolineò in dettaglio il processo di formazione dell’urina utilizzando la tecnica di micropuntura fatta nel rene di rana. Richards collaborò già con il Merck Institute su richiesta di George Merck, fondatore della casa farmaceutica Merck & Co.
agenti organomercuriali
I primi diuretici sintetici dei primi anni del XX secolo erano gli agenti organomercuriali. Questi furono ampiamente usati negli anni quaranta, specialmente in aggiunta alla terapia dell’insufficienza cardiaca congestizia. Fu ipotizzata l’aggiunta di cloruro d’ammonio per aumentare l’effetto diuretico della mercupurina e della mercuidrina, così era il nome commerciale di questi organomercuriali in USA, ma tale ipotesi non venne mai applicata nella pratica per i continui reperti addominali che rivelavano la presenza di compresse di cloruro d’ammonio inassorbito nel tratto intestinale. Si riteneva che gli agenti organomercuriali esercitavano la loro azione diretta bloccando le deidrogenasi sulfidril-catalizzate rendendo così disponibili gli idrogenioni per lo scambio con i sodioioni. Sebbene efficaci come diuretici, tali composti potevano essere somministrati solo per via parenterale, il che costituiva un inconveniente nella terapia cronica.
INIBITORI DELL’ANIDRASI CARBONICA
Nel 1940, Mann e Keilin dimostrarono che la sulfanilamide era un inibitore specifico dell’anidrasi carbonica. Cinque anni dopo, nel 1945, Pitts e Alexander chiarirono il ruolo dell’anidrasi carbonica nell’acidificazione delle urine a livello del tubulo renale. Il report di Krebs del 1948 confermò che i sulfonamidi possedevano in generale la proprietà di inibire l’anidrasi carbonica. Tali report portarono Schwartz ad investigare gli effetti della sulfanilamide sull’escrezione di sale ed acqua nell’insufficienza cardiaca congestizia. Nel 1949 egli propose la sulfanilamide come debole agente natriuretico-diuretico.
Nello stesso tempo, certe case farmaceutiche diressero le loro ricerche chimiche verso la sintesi di congeneri della sulfonamide per l’inibizione dell’anidrasi carbonica.
Nel 1950, due ricercatori dei laboratori Lederle, Roblin e Clapp, riportarono i loro risultati con certi inibitori. Il DIAMOX (acetazolamide), fu uno di quelli, e come risultato del report di Maren nel 1952 divenne un popolare agente orale diuretico. Comunque, il suo effetto diuretico era meno potente di quello degli organomercuriali.
Venne studiato in questo periodo un altro percorso della diuresi. Questo si basava sulla semplice nozione che se si ostacolava il riassorbimento tubulare degli ioni sodio, la proprietà osmotica intrinseca di tale ione avrebbe determinato l’eliminazione di una certa quantità di urina. Lo scopo era dunque quello di trovare un congenere dei sulfonamidi che fosse un agente saluretico. Il DIAMOX non apparteneva a questa categoria. I chimici della Sharp & Dohme furono attivi nel ricercare questo tipo di agente.
Il farmacologo statunitense Karl Henry Beyer jr (19giugno 1914 – 2dicembre 1996) guidava questo gruppo. Al simposio della International Physiology Congress del 1953, Beyer presentò un nuovo inibitore dell’anidrasi carbonica derivato dalla famiglia dei sulfonamidi che aveva in aggiunta un’azione saluretica. Questo era il p-carbossibenzenesulfonamide (CBS). Esso non raggiunse mai nessun successo, probabilmente a causa della popolarità del DIAMOX.
TIAZIDICI
Ad ogni modo, Beyer ed i suoi colleghi continuarono la loro ricerca di un saluretico utile dal punto di vista clinico e nel 1958 riuscirono a pubblicare i loro primi report sul clorotiazide. Questo fu il primo sulfonamide saluretico attivo con la somministrazione orale. In poco tempo i tiazidi divennero un importante componente della terapia diuretica. Il clorotiazide fu commerciato con il nome DIURIL e l’idroclorotiazide come HYDRODIURIL.
DIURETICI DELL’ANSA
I cosiddetti diuretici dell’ansa entrarono in campo negli anni sessanta del novecento. Essi sono in realtà agenti cloruretici. I primi furono la furosemide e l’acido etacrinico. Più recentemente è stata introdotta la bumetanide per opera di Duchin e Davies che stavano lavorando in maniera indipendente tra di loro. I diuretici dell’ansa furono accettati in maniera entusiastica dai clinici non solo perché potevano essere somministrati sia per via orale che parenterale, ma anche perché possedevano una potente capacità diuretica. Comunque, questi nuovi composti dimostrarono di avere un marcato effetto kaliuretico, il che costituì un significativo difetto clinico dal momento che l’ipopotassiemia predispone alle aritmie cardiache. Nello sforzo di aggirare tale ostacolo furono introdotti, sempre negli anni sessanta, l’amiloride ed il triamterene.
ANTIALDOSTERONICI
L’antagonismo dell’aldosterone è un altro interessante percorso della diuresi che venne esplorato per la prima volta negli anni cinquanta del novecento.
Il ruolo fisiologico dell’aldosterone fu svelato nel 1954 grazie agli sforzi della biochimica inglese Sylvia Agnes Sophia Wardropper Simpson Tait (8gennaio 1917 – 28febbraio 2003 – Usò prima il cognome del primo marito, Simpson, e poi quello del secondo, Tait), suo marito James Francis Tait (1926-2014) e colleghi. Questo ormone della corteccia surrenale causa la ritenzione di sodio e l’escrezione di potassio. Ovviamente, un antagonista di questo meccanismo sembra essere un farmaco adatto ad eliminare il sodio e determinare una conseguente diuresi.
Il primo agente con proprietà antialdosteroniche era lo spironolattone. Questo fu descritto da C. M. Kagawa ed i suoi associati dei Laboratori Searle nel 1957 sulla rivista Science.
Prima del 1950 la lista dei rimedi per controllare l’elevata pressione sanguigna fu sbalorditiva nonostante fossero inutili e talvolta addirittura tossici. Questi passavano dagli estratti botanici alle diete di riso, dalla restrizione di sodio alla nefrotomia, e così via. La lista è troppo lunga per enumerare tutti i rimedi. La simpatectomia di Smithwick sembrò essere il solo valido approccio ma questo richiedeva un intervento chirurgico.
ANTITROMBOTICI
L’uso degli anticoagulanti in medicina non è nuovo. Le sanguisughe, con la loro capacità di estrarre il sangue liberamente e senza formazione di coaguli, sono state per più di mille anni una valida alternativa al salasso attraverso venesezione. Haycraft confermò questa capacità anticoagulante delle sanguisughe nel 1884 con le sue osservazioni in materia. Prima di tutto, egli notò che il morso di una sanguisuga determinava lo scorrimento del sangue senza formazione del coagulo. Inoltre, egli scoprì che il sangue della comune sanguisuga, la “hirudo medicinales”, un verme anellide, non coagulava nemmeno dopo essere stato estratto dall’animale. Il principio attivo anticoagulante di questo anticoagulante nel sangue fu isolato da Jacoby nel 1904. Egli lo estrasse dalla stesso tipo di sanguisuga e gli diede logicamente il nome di “irudina”.
EPARINA
L’eparina fu scoperta all’inizio del novecento. Due uomini furono i protagonisti principali della sua scoperta: Howell e McLean. C’è una certa controversia sul ruolo che questi due svolsero nella sua scoperta e sviluppo. William Henry Howell seguì i suoi studi pre-medici alla Johns Hopkins University. Sebbene relativamente nuova, la scuola si era già creata l’immagine di un istituto che promuove la ricerca e che non aveva paragone con le altre università americane. Howell lavorò nel dipartimento di biologia di Newell Martin. Il maestro assegnò ad Howell il ruolo di investigare la differenza di coagulazione tra il sangue venoso e quello arterioso. Egli passò non più di due mesi su questo progetto prima di metter da parte il lavoro per cercare l’origine della fibrina formata nella coagulazione del sangue. Nel corso del tempo egli cercò di dimostrare che il sangue dei mammiferi contiene inibitori fisiologici della coagulazione, e nel 1911 provò la presenza di una sostanza di questo tipo che la chiamò antitrombina.
Le ricerche di Howell costituirono la base per l’isolamento dell’eparina nel 1916 da parte di Jay McLean, uno studente di medicina californiano, che allora lavorava nel laboratorio di Howell. Howell aveva proposto a McLean il problema di determinare il significato ed il ruolo della coagulazione. Fu durante il corso di questo lavoro che McLean scoprì l’anticoagulante. Agendo contro i desideri del suo mentore, il giovane studente si precipitò a stampare i risultati della sua ricerca. Egli la chiamò cefalina. La sostanza era impura e molto tossica. Ad Howell fu lasciato il compito di preparare una forma più purificata. Dopo due anni dalla scoperta di McLean, Howell era già totalmente immerso in questa nuova ricerca. Nel 1918 Howell descrisse l’anticoagulante in maniera più dettagliata e la chiamò eparina al posto del termine antiprotrombina inizialmente suggerito.
Una dettagliata descrizione del suo comportamento chimico e fisiologico fu presentata da Howell nel 1928. In questo periodo la capacità dell’eparina di impedire la coagulazione intravascolare fu ampiamente dimostrata in numerosi esperimenti animali. L’interesse di Charles Best verso l’eparina lo portò a diventare colui che per primo la somministrò sull’uomo nel 1935. Tre anni dopo Irving Wright, della Cornell University, rivolse la sua attenzione verso questa sostanza allorchè egli stesso cadde vittima di una tromboflebite. Il valore profilattico dell’eparina nella trombosi post-chirurgica fu dimostrato da Clarence Crafoord nel 1937.
Un grande limite dell’eparina è sempre stata l’impossibilità di somministrarla oralmente. È per questa ragione che i derivati coumarinici divennero un’alternativa all’eparina nelle diverse indicazioni che richiedevano terapia prolungata o cronica con anticoagulanti.
Dicumarolici
La storia del dicumarolo inizia nei primi anni venti del novecento in quella regione del Nord America in cui si trovano il Nord Dakota ed Alberta. I bestiami venivano decimati da un particolare disordine che era caratterizzato da emorragia. Due veterinari, Schofield e Roderick, implicarono il “melilotus officinalis” come la causa dell’epidemia. Il disturbo divenne noto come malattia del “melilotus officinalis”. Non si scoprì nient’altro sulla materia fino al 1933, anno in cui Karl Paul Link si confrontò con un contadino sconvolto dalla morte di una vacca ricoperta di ematomi. L’agricoltore era disperato e chiese aiuto a Link per cercare di capire in che modo il “melilotus officinalis” riusciva a provocare tale scempio nel bestiame. Quella notte nel Wisconsin c’era stata fu una bufera di neve ed il contadino aveva nutrito la sua vacca con del “melilotus officinalis” dal momento che non aveva a disposizione nessun altro alimento a causa del brutto tempo. Link raccolse la sfida e dovettero passare altri sette anni prima che scoprisse la causa della diatesi emorragica di quegli sfortunati armenti che si cibavano Link raccolse la sfida e dovettero passare altri sette anni prima che scoprisse la causa della diatesi emorragica di quegli sfortunati armenti che si cibavano di “melilotus officinalis”. Nel 1940 egli identificò l’agente causale nel dicumarolo. Egli riuscì anche a sintetizzare tale agente e, nel corso dei due anni successivi, il suo laboratorio alla Agricultural Experimental Station riuscì a sintetizzare più di 100 idrocumarinici collegati. Questi furono elencati secondo un ordine numerico a seconda della loro struttura chimica di base. Quelli con il numero tra il 40 ed il 65 furono studiati in maniera più dettagliata per le loro percepite potenzialità anticoagulanti. Al di fuori di questo gruppo stavano due agenti molto potenti quando somministrati al topo o al cane. Egli selezionò il numero 42 come prodotto ideale per uccidere i topi e gli diede il nome warfarin. Warfarin è un acronimo in cui “warf” deriva dalle prime lettere di Wisconsin Alumnae Research Foundation (che finanziò la ricerca) e “arin” da coumarin. Il warfarin divenne disponibile per l’uso clinico dopo le indicazioni di Samuel M. Gordon e i Laboratori Endo, i quali erano stati invitati a loro volta a studiare tale agente da Karl Link. La Endo lo mise in commercio con il nome COUMADIN.
Trial multicentrici furono avviati negli Stati Uniti sotto l’energetica guida di Irving Wright nel 1948. Tale studio generò così tanto entusiasmo per quasi due decadi che per un medico era praticamente impossibile astenersi da tale approccio nel trattamento dell’infarto miocardico. Successivi studi clinici gettarono acqua sul fuoco su tale approccio ed alla fine venne sconfessato come adeguata misura routinaria. Il fatto era che nessuno di questi trial clinici aveva dimostrato in maniera irrefutabile che questo trattamento nell’infarto miocardico abbassava la mortalità.
Il tempo di protrombina fu scoperto dall’ematologo statunitense Armand James Quick (1894 – 26gennaio 1978) e colleghi nel 1935. L’INR (International Normalized Ratio) è stato inventato all’inizio degli anni ’80 del novecento da Tom Kirkwood mentre lavorava alla “UK National Institute for Biological Standards and Control” per fornire un modo coerente di esprimere il rapporto del tempo di protrombina, che aveva precedentemente sofferto di un ampio grado di variazione tra i centri che utilizzano reagenti diversi.
TERAPIA TROMBOLITICA
L’avvento della terapia trombolitica nella gestione in emergenza dell’infarto miocardico acuto e l’angina instabile ha creato una ripresa d’interesse verso l’anticoagulazione come misura aggiuntiva dopo la trombolisi efficace.
Nel 1947 Tage Astrup e Permin individuarono in molti tessuti animali un agente in grado di attivare il plasminogeno; questo fattore, inizialmente chiamato fibrochinasi, è oggi conosciuto come attivatore tissutale del plasminogeno (t-PA). Allo stesso periodo risale la dimostrazione dell’esistenza di un attivatore del plasminogeno nelle urine da parte di due ricercatori di Oxford, R. G. MacFarlane e J. Pilling, successivamente denominato urochinasi.
Sol Sherry, che lavorava alla Washington University di St Louis, insieme ad Anthony Fletcher e Norma Alkjaersig, applicò la sua conoscenza di enzimologia batterica agli eventi vascolari trombotici. Egli dimostrò che i coaguli sanguigni potevano essere dissolti in vivo con la streptochinasi.
Dopo gli anni settanta, in seguito al boom delle biotecnologie creato da Cohen e Boyer, si è assistito ad una vera e propria esplosione della ricerca applicato al campo del DNA ricombinante e a nuove acquisizioni nel campo della fibrinolisi; basti pensare al lancio dopo l’urochinasi e la streptochinasi di due farmaci fibrinolitici costruiti tramite tecniche biotecnologiche, l’alteplase e tenecteplase (approvato dalla FDA nel 2000), indicati per il trattamento fibrinolitico dell’embolia polmonare e dell’infarto miocardico acuto con sopraslivellamento del tratto ST entro le prime ore dall’insorgenza dei sintomi.
antiaggreganti
Negli anni ottanta del XX secolo gli agenti antipiastrinici sono diventati parte integrante dell’armamentario antitrombotico. Ciò è avvenuto perché la malattia arteriosa coronarica è spesso precipitata dall’aggregazione piastrinica. Tali agenti includono l’aspirina, il dipiridamolo e il sulfinpirazone.
L’efficacia dell’aspirina come agente antipiastrinico è stata riconosciuta al National Heart, Lung and Blood Institute Consensus Conference sulla terapia antitrombotica. L’aspirina (o acido acetilsalicilico o ASA; nome IUPAC: acido 2-acetossibenzoico) è un farmaco antinfiammatorio non-steroideo (FANS) della famiglia dei salicilati. Il suo numero CAS è 50-78-2.La sostanza attiva dell’estratto di corteccia del salice bianco (Salix alba), chiamato salicina, fu isolata in cristalli nel 1828 da Henri Leroux, un farmacista francese, e da Raffaele Piria, un chimico italiano. Il composto venne isolato anche dai fiori di olmaria (Spiraea ulmaria) da alcuni ricercatori tedeschi nel 1839.
Nel 1897 Felix Hoffmann (21gennaio 1868 – 8febbraio 1946)su l’idea del suo superiore Arthur Eichengrün, chimici impiegati presso la Friedrich Bayer & Co., derivò il gruppo ossidrile (-OH) dell’acido salicilico con un gruppo acetile, formando l’acido acetil-salicilico. Tale composto presentava gli stessi effetti terapeutici dell’acido salicilico, ma con minori effetti collaterali. Nacque così il primo farmaco sintetico – una molecola nuova, non una copia di una molecola già esistente in natura – e la moderna industria farmaceutica. Il nome “aspirina” fu brevettato dalla Bayer il 6marzo 1899, componendo il prefisso “a-” (per il gruppo acetile) con “-spir-” (dal fiore Spiraea, da cui si ricava l’acido spireico, ovvero l’acido salicilico) e col suffisso “-ina” (generalmente usato per i farmaci all’epoca). La Bayer perse il diritto ad usare il proprio marchio in molte nazioni, dopo che gli Alleati occuparono e rivendettero le sue proprietà in seguito alla prima guerra mondiale. In una ricerca che gli valse un premio Nobel, il londinese John Vane dimostrò che l’aspirina nell’organismo umano blocca la produzione delle prostaglandine e dei trombossani.
Si racconta che il chimico che per primo scoprì gli effetti dell’aspirina sia stato non Felix Hoffmann ma Arthur Eichengrün e che il suo nome sia stato escluso dalla storia ufficiale della Bayer nel 1934 in quanto scienziato ebreo e dunque non gradito al regime nazista.
ANTIARITMICI
Prima del 1950 l’unico farmaco antiaritmico disponibile era la quinidina. Questa si distingue dagli altri anche perché è il solo agente di origine botanica. Essa è imparentata con la quinina che a sua volta deriva dalla corteccia di china. La quinidina come la digitale è un sopravvissuto. Nonostante le sue tante reazioni avverse essa è ancora un antiaritmico largamente usato.
Nel 1950 comparvero due farmaci che entrarono con successo in competizione con la quintina all’attenzione dei cardiologi: la procainamide e la lidocaina. Entrambi erano degli anestetici locali. Gli anni sessanta videro l’introduzione del disopiramide, dei beta-bloccanti, calcio antagonisti e bloccanti dei canali lenti del potassio.
AMIODARONE
L’osservazione che la molecola progenitrice dell’amiodarone, la chellina, avesse proprietà cardioattive, fu riportata dal fisiologo russo Gleb von Anrep (23settembre 1891 – 11gennaio 1955), mentre lavorava al Cairo, nel 1945.
In seguito alla scoperta nel 1961 da parte di R. Tondeur e F. Binon, due chimici belgi che stavano lavorando su preparati derivati dalla chellina presso l’industria farmaceutica Labaz di Bruxelles, l’amiodarone venne usato soprattutto in Europa come anti-anginoso.
In seguito all’esperienza e alle ricerche del Dr. Bramah N. Singh (3marzo 1938 – 20settembre 2014), Università di Oxford, riportate nel 1970, venne riconsiderato il ruolo dell’amiodarone che, unitamente al sotalolo, venne classificato come farmaco anti-aritmico di classe III, gruppo caratterizzato dalla capacità di allungare il periodo refrattario mediante il blocco dei canali del potassio. Singh era un allievo di Miles Vaughan Williams (8agosto 1918 – 31agosto 2016), il farmacologo britannico meglio conosciuto per la “classificazione Vaughan Williams dei farmaci antiaritmici” introdotta nel 1970.
Il Dr. Mauricio Rosenbaum (20 agosto 1921 – 4maggio 2003), medico argentino, basandosi sui lavori del Dr. Singh, iniziò ad usare negli anni settanta l’amiodarone per il trattamento dei pazienti che soffrivano di aritmia sopraventricolare e aritmia ventricolare, con risultati impressionanti. In accordo con i risultati di Singh e Rosenbaum, i medici americani cominciarono ad usare l’amiodarone come terapia cronica nei pazienti aritmici a partire dal 1970. Dal 1980 l’amiodarone cominciò ad essere prescritto anche negli stati europei.
In seguito ai gravi effetti collaterali polmonari, cardiaci e tiroidei (associati all’uso cronico), venne riconsiderato l’impiego clinico dell’amiodarone nelle tachiaritmie.
Nel dicembre 1985 l’amiodarone venne approvato dalla Food and Drug Administration per il trattamento della fibrillazione atriale e nella profilassi della tachicardia ventricolare ricorrente.
IPOCOLESTEROLEMIZZANTI
La gestione farmacologica dell’arteriosclerosi ha dovuto attendere il chiarimento del ruolo del colesterolo nella sua patogenesi. Ciò è avvenuto solo nel XX secolo. Lo sviluppo di agenti che riducono i livelli ematici di colesterolo è iniziata nella seconda parte di questo secolo. I primi preparati agivano solo a livello intestinale impedendo l’assorbimento del colesterolo ingerito attraverso l’azione di legame delle resine.
Negli anni sessanta del novecento si cercò di ridurre i livelli ematici del colesterolo bloccando la sua sintesi cellulare. Sebbene l’idea fosse buona (MER 29), il blocco della sintesi a livello del suo penultimo precursore ebbe disastrose conseguenze.
Nel 1971, Akira Endo, un biochimico giapponese che lavorava presso la compagnia farmaceutica Sankyo, iniziò la ricerca di farmaci ipocolesterolemizzanti. La ricerca aveva già mostrato che il colesterolo viene prodotto nel fegato tramite l’azione di un enzima noto con il nome HMG-CoA reduttasi. Endo ed il suo team riuscirono nell’intento di far produrre da certi microrganismi inibitori dell’enzima, come il mevalonato, un prototipo di alcune statine. La prima statina prodotta da Endo fu la mevastatina, molecola prodotta dal fungo “Penicillum Citrinum”, ma questa non venne mai messa in commercio a causa dei numerosi effetti collaterali.
Negli anni ottanta la lovastatina venne introdotta coma inibitore della CoA-redattasi da parte della compagnia farmaceutica americana Merck. Tale farmaco ebbe successo semplicemente perché era molto efficace nel bloccare la sintesi epatica del colesterolo nelle sue prime fasi senza molti effetti collaterali. Negli anni novanta sono iniziati a comparire report che mostrano la regressione dell’aterosclerosi grazie alla riduzione del colesterolo sierico.
Blankenhorn ha svolto un importante ruolo nel descrivere questi effetti benefici attraverso documentazione angiografica della regressione.
riferimenti:
- Testo “The History of Cardiology” del prof. Louis J. Acierno (cap. 30)
- Cecilia C. Mettler & Fred A. Mettler: “History of Medicine“, 1947
- https://en.wikipedia.org/wiki/Cecilia_Mettler
- https://it.wikipedia.org/wiki/Matteo_Plateario
- Part II – Anonymity in the Printed English Herbal (Published online by Cambridge University Press: 23 December 2021)
- https://en.wikipedia.org/wiki/Grete_Herball
- https://en.wikipedia.org/wiki/James_Hope_(physician)
- https://it.wikipedia.org/wiki/Ippolito_Francesco_Albertini
- https://en.wikipedia.org/wiki/Samuel_Hahnemann
- https://en.wikipedia.org/wiki/Christian_Friedrich_Sch%C3%B6nbein
- https://en.wikipedia.org/wiki/Constantine_Hering
- https://en.wikipedia.org/wiki/William_Murrell_(physician)
- https://it.wikipedia.org/wiki/Eugene_Braunwald
- https://it.wikipedia.org/wiki/John_Jacob_Abel
- https://en.wikipedia.org/wiki/Takamine_J%C5%8Dkichi
- https://en.wikipedia.org/wiki/Albrecht_Fleckenstein
- https://it.wikipedia.org/wiki/James_W._Black
- https://en.wikipedia.org/wiki/Alfred_Newton_Richards
- http://www.nasonline.org/publications/biographical-memoirs/memoir-pdfs/beyer-karl-jr.pdf
- Kurze Mitteilungen Published: 01 March 1954 “Konstitution des Aldosterons, des neuen Mineralocorticoids“, di S. A. Simpson, J. F. Tait, A. Wettstein, R. Neher, J. v. Euw, O. Schindler & T. Reichstein
- https://en.wikipedia.org/wiki/Sylvia_Agnes_Sophia_Tait
- https://en.wikipedia.org/wiki/James_Francis_Tait
- “Action of New Steroids in Blocking Effects of Aldosterone and Deoxycorticosterone on Salt“, di C. M. KAGAWA, J. A. CELLA, AND C. G. VAN ARMAN – Authors Info & Affiliations (SCIENCE; 15 Nov 1957; Vol 126, Issue 3281)
- https://en.wikipedia.org/wiki/Armand_J._Quick
- https://en.wikipedia.org/wiki/Prothrombin_time#History
- “Fibrinolytic Activity of Normal Urine“, di Macfarlane, R. G. (Pathological Department, Radcliffe Infirmary, Oxford); Pilling, J. – Nature, Volume 159, Issue 4049, pp. 779 (june 1947).
- https://en.wikipedia.org/wiki/Urokinase#
- https://en.wikipedia.org/wiki/Streptokinase#History
- https://it.wikipedia.org/wiki/Amiodarone#Storia
- “AMMI VISNAGA IN THE TREATMENT OF THE ANGINAL SYNDROME*“, BY GLEB VON ANREP, G. S. BARSOUM, M. R. KENAWY, AND G. MISRAHY (From the Physiological Laboratory and the University Hospital, Cairo, Egypt Received August 28, 1945)
- https://en.wikipedia.org/wiki/Amiodarone#History
- “The effect of amiodarone, a new anti-anginal drug, on cardiac muscle“, di B. N. SINGH* AND E. M. VAUGHAN WILLIAMS (Department of Pharmacology, University of Oxford) – Br. J. Pharmac. (1970), 39, 657-667
- https://en.wikipedia.org/wiki/Bramah_N._Singh
- https://en.wikipedia.org/wiki/Miles_Vaughan_Williams
- https://en.wikipedia.org/wiki/Antiarrhythmic_agent#Vaughan_Williams_classification
- https://www.revespcardiol.org/en-mauricio-b-rosenbaum-last-all-round-articulo-13052421
- “Clinical efficacy of amiodarone as an antiarrhythmic agent“, di Mauricio B. Rosenbaum, MD, FACC; Pablo A. Chiale, MD; M. Susana Halpern, MD; Raúl J. Levi, MD; Julio O. Lázzari, MD; Marcelo V. Elizari, MD, FACC (DOI:https://doi.org/10.1016/0002-9149(76)90807-9); REPORT ON THERAPY| VOLUME 38, ISSUE 7, P934-944, DECEMBER 1976
- https://en.wikipedia.org/wiki/Akira_Endo_(biochemist)
- Paolo Larizza, “Trattato delle malattie del Sangue” (Piccin Editore, 1991), capitolo 69 (fibrinolisi)
Autore: dott. Concetto De Luca (novembre 2011)





































