La sindrome da deplezione del DNA mitocondriale: breve cronistoria
- articolo del prof. Sergio Barocci
Il caso della piccola Indi Gregory
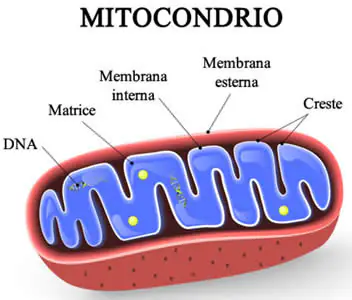
Con sindrome da deplezione del DNA mitocondriale (MDS) si identificano diversi disturbi genetici rari, in particolare quella che ha colpito la piccola inglese Indi Gregory per la quale si conoscono pochissimi casi (50 casi al mondo) caratterizzati dalla riduzione del numero di mitocondri all’interno delle cellule. Il numero dei mitocondri varia nell’organismo a seconda del tipo cellulare; l’epatocita contiene fino a duemila mitocondri, mentre il glubulo rosso ne è praticamente privo.
Il DNA mitocondriale ha, con molta probabilità, un’origine batterica. Infatti, sulla base di numerosi studi indipendenti, i biologi molecolari ritengono che la presenza cellulare del DNA mitocondriale sia il risultato dell’incorporazione, da parte delle cellule eucariotiche ancestrali, di organismi batterici indipendenti, molto simili ai mitocondri.
DNA e catena respiratoria MITOCONDRIALE

Quasi tutto il nostro DNA è localizzato all’interno del nucleo, in strutture chiamate cromosomi. Una piccola parte è però presente nei mitocondri, piccoli organelli all’interno della cellula a forma ovoidale lunghi circa 2 micron che funzionano come delle centrali energetiche della cellula.
La lunghezza totale del DNA nucleare è circa 3 miliardi di nucleotidi mentre quella del DNA mitocondriale è solo 16.500 nucleotidi. il DNA mitocondriale è una molecola circolare, mentre il DNA nucleare è una molecola lineare.
Anche se di piccole dimensioni, il DNA mitocondriale è estremamente importante per le nostre cellule perché è necessario per far funzionare la catena respiratoria mitocondriale (avviene attraverso le fasi rispettivamente della glicolisi, della decarbossilazione ossidativa del piruvato, del ciclo di Krebs e della fosforilazione ossidativa), il sistema che l’energia chimica contenuta nel cibo trasforma in ATP (Adenosina trifosfato), ossia la principale fonte di energia sfruttabile dalla cellule.
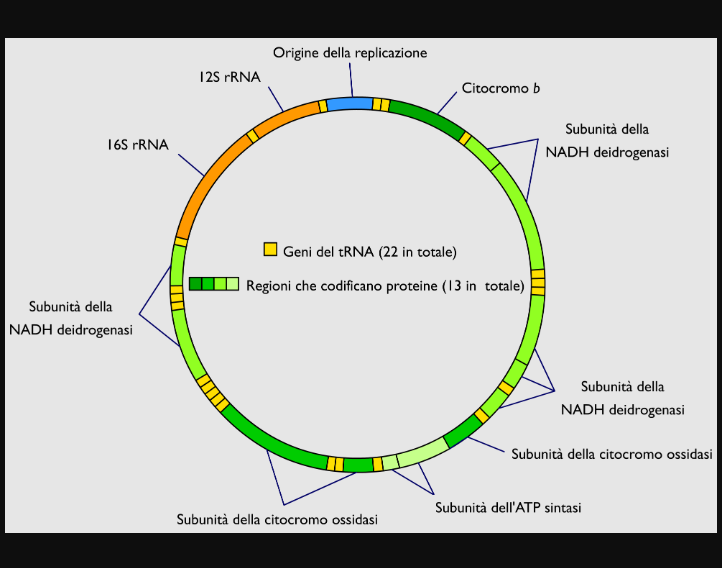
Geni del DNA mitocondriale
{kind=link}
Il DNA mitocondriale è stato scoperto all’inizio degli anni sessanta dai coniugi Margit MK Nass-Edelson ed il marito Sylvan Nass (Istituto di Biologia Sperimentale dell’Università di Stoccolma) mediante microscopia elettronica come “fibre sensibili alla DNasi” all’interno di mitocondri di embrione di pollo, e da Ellen Haslbrunner, Hans Tuppy e Gottfried Schatz (Istituto di Biochimica dell’Università di Vienna) mediante analisi biochimiche su frazioni mitocondriali altamente purificate.
I geni del DNA mitocondriale sono in numero di 37 e codificano per proteine, RNA transfer (tRNA) e RNA ribosomiale (rRNA). Il DNA mitocondriale possiede pochissime sequenze di nucleotidi non codificanti, cioè che non producono alcuna proteina, tRNA o rRNA. In termini percentuali, soltanto il 3% del DNA mitocondriale è non codificante.
All’opposto, il DNA nucleare è codificante soltanto per il 7%. Per funzionare adeguatamente, Il DNA mitocondriale ha bisogno del supporto di alcuni prodotti genici, derivanti dal DNA nucleare.
37 geni
Esattamente:
• 13 geni per 13 proteine deputate alla realizzazione della fosforilazione ossidativa
• 22 geni per 22 molecole di tRNA
• 2 geni per due molecole di rRNA
Il DNA mitocondriale viene trasmesso esclusivamente dalla cellula uovo della madre, in quanto gli spermatozoi non forniscono infatti alcun mitocondrio all’atto della fecondazione. Tale scoperta venne fatta nel 1980 da scienziati del Dipartimento di Genetica e Pediatria dalla Stanford University Medical School, in California.
sindrome da deplezione del DNA mitocondriale
I mitocondri, nelle sindromi da deplezione del DNA mitocondriale, sono incapaci di produrre energia a sufficienza, con effetti devastanti sull’organismo nel complesso e in particolar modo sugli organi che consumano molta energia, come cervello, cuore, muscoli e fegato.
Inoltre, queste sindromi pur essendo molto eterogenee si riescono a distinguere, per la loro manifestazione precoce, nei primi giorni o mesi di vita e, in molti casi, per il rapido aggravarsi dei sintomi.
Quella di cui è affetta Indi Gregory è una delle forme più aggressive e precocemente letali. In particolare, si chiama aciduria combinata D,L-2-idrossiglutarica, è associata con elevati livelli di D-2 idrossiglutarato e L-2 idrossiglutarato, ed il gene responsabile è lo SLC25A1 (solute carrier family 25 member 1).
SLC25A1 di trasporto mitocondriale del citrato
Il SLC25A1 di trasporto mitocondriale del citrato, noto anche come proteina trasportatrice del citrato (CTP) o trasportatore citrato/isocitrato (CIC), è un trasportatore di membrana mitocondriale codificato nel nucleo dal gene SLC25A1 situato sul cromosoma 22. Ad oggi, rappresenta l’unico trasportatore mitocondriale umano conosciuto per il citrato, il che rende la sua attività di fondamentale importanza.
La funzione nota di questo trasportatore consiste nel promuovere il trasporto di acido citrico o acido isocitrico dai mitocondri nel citosol in cambio dell’ acido malico. L’acido citrico citosolico ha diverse funzioni fondamentali, da un lato di fornire la fonte per l’acetil-coenzima A (Ac-CoA) necessaria per la biosintesi degli acidi grassi e degli steroli, e dall’altro, fungendo da regolatore allosterico degli enzimi che controllano la glicolisi, la lipogenesi e la gluconeogenesi
Acido Citrico
L’attività della fosfofruttochinasi (PFK), che è un enzima glicolitico, viene inibita dal legame con l’acido citrico o citrato, mentre l’1,6-bisfosfatasi (FBP1) e l’acetil-CoA carbossilasi alfa (ACACA), necessarie rispettivamente per la gluconeogenesi e la sintesi lipidica, vengono attivate dall’acido citrico. E’ stato evidenziato che attraverso l’attività di importazione inversa, il trasportatore del citrato può anche promuovere l’ingresso del citrato citosolico nei mitocondri, stimolando il ciclo dell’acido tricarbossilico e la fosforilazione ossidativa e mantenendo l’equilibrio redox attraverso la generazione di NADPH, tutte attività sono importanti per l’espansione delle cellule staminali/iniziatrici del cancro e per la crescita indipendente dall’ancoraggio delle cellule tumorali
Inoltre, il trasportatore del citrato è probabilmente anche un mediatore del “cross-talk mitocondriale-nucleo” attraverso il quale gli aggiustamenti metabolici originati dai mitocondri vengono trasmessi al nucleo e rimodellano il programma di trascrizione attraverso la regolazione epigenetica. Questa attività del trasportatore deriverebbe dalla sua capacità di fornire Ac-CoA per le reazioni di acetilazione, di migliorare la disponibilità di intermedi del ciclo dell’ ac. tricarbossilico che agiscono come regolatori epigenetici, in particolare succinato, fumarato e alfa-chetoglutarato (α-KG), e di prevenire l’accumulo di acidi l- e D-2-idrossiglutarico, due oncometaboliti che inibiscono le demetilasi istoniche e sono anormalmente elevati quando l’attività del trasportatore è compromessa.
aciduria combinata D,L-2-idrossiglutarica
Dunque, il gene SLC25A1 codifica per un membro della sottofamiglia dei trasportatori mitocondriali delle proteine trasportatrici dei soluti. I membri di questa famiglia includono trasportatori codificati nel nucleo che traslocano piccoli metaboliti attraverso la membrana mitocondriale. Il gene in questione è specializzato nel trasporto del citrato, composto organico fondamentale per produrre ATP che come già riferito fornisce alla cellula l’energia necessaria per funzionare. In questa mutazione esso non compie la funzione dovuta e crea la malattia. La malattia si trasmette se vengono ereditate due copie del gene mutato da entrambi i genitori.
Essa, in passato era nota per altre cause genetiche, mentre la particolare forma rara di cui soffre Indi Gregory è stata scoperta soltanto nel 2013.
FORMA CLINICA DELL’aciduria combinata D,L-2-idrossiglutarica
Si tratta di una forma di encefalopatia grave con epilessia, una malattia progressiva, che nei casi più gravi si manifesta fin dalla nascita con crisi epilettiche, insufficienza respiratoria, ritardo dello sviluppo e malformazioni, come quelle che impediscono la formazione del corpo calloso che collega i due emisferi del cervello o la formazione del nervo ottico.
Nei casi più lievi provoca invece debolezza dei muscoli, soprattutto di braccia e gambe.
Non esiste al momento una cura, se non forme palliative per alleviare il dolore come quelle che sarebbero state messe in atto al Centro di cure palliative di Passoscuro dell’Ospedale pediatrico Bambino Gesù di Roma.
La soluzione potrebbe essere individuata solo nella terapia genica ed in farmaci a tecnologia RNA, ma al momento i test per queste terapie innovative sono solo in fase iniziale.
varianti di sindrome da deplezione del DNA mitocondriale
Esistono alcune varianti di sindrome da deplezione del DNA mitocondriale, differenti fra loro per sintomatologia, organi interessati, durata della sopravvivenza e rapidità nella progressione clinica
Le patologie si manifestano tutte nei primissimi giorni di vita, o comunque durante l’infanzia, e nella stragrande maggioranza dei casi è proprio in questa fase, entro i due anni, che i bambini perdono la vita. I malati di alcune forme di sindrome da deplezione del DNA mitocondriale possono sopravvivere fino all’adolescenza, e in rarissimi casi giungere all’età adulta.
tre forme principali DI MDS
Le tre forme principali di sindrome da deplezione del DNA mitocondriale sono la miopatica, che colpisce i muscoli scheletrici; l’epatopatica che interessa il fegato e l’encefalomiopatica, che riguarda muscoli e cervello.
Per quanto concerne le cause, la sindrome da deplezione del DNA mitocondriale è legata alle mutazioni anche di altri diversi geni come il TK2 (thymidine kinase 2) nella forma miopatica, il SUCLA2 ( succinate-CoA ligase ADP-forming subunit beta) in quella encefalomiopatica con aciduria metil-malonica e DGUOK (deoxyguanosine kinase), POLG (DNA polymerase gamma, catalytic subunit) e MPV17 (mitochondrial inner membrane protein MPV17 ) per quella epatopatica.
BREVE CRONISTORIA

La storia delle “patologie mitocondriali”, cioè delle patologie genetiche che colpiscono il metabolismo mitocondriale a causa di mutazioni nei geni codificati dal DNA nucleare per proteine attive all’interno dei mitocondri o da mutazioni nei geni codificati dal DNA mitocondriale, inizia nel 1988.
In quell’anno, due diversi gruppi di ricercatori scoprirono, rispettivamente, singole delezioni su larga scala del DNA mitocondriale (mtDNA) nelle biopsie muscolari di pazienti con “miopatie mitocondriali” e una mutazione puntiforme nel gene mtDNA per la subunità 4 della NADH deidrogenasi ( MTND4 ), associata all’ottica ereditaria di Leber ereditata per via materna neuropatia (LHON).
UNA NUOVA GENETICA MITOOCONDRIALE
Da allora in poi, nacque una nuova “genetica mitocondriale” concettuale, separata dalla genetica mendeliana, basata su tre caratteristiche del mtDNA: (1) poliplasmia; (2) eredità materna; e (3) segregazione mitotica. La diagnosi delle malattie correlate al mtDNA è diventata possibile attraverso l’analisi genetica e approcci sperimentali che comportano la colorazione istochimica di sezioni muscolari o cerebrali, la reazione a catena della polimerasi a fibra singola (PCR) del mtDNA e la creazione di “cibridi” (ibridi citoplasmatici) immortali derivati dal paziente, linee cellulari di fibroblasti.
La disponibilità delle tecniche sopra menzionate insieme alla nuova sensibilità dei medici verso tali disturbi hanno portato alla caratterizzazione di un numero in costante crescita di patologie.
IL 1988

Due grandi scoperte furono fatte nel 1988 da due famosi ricercatori, Anita Harding presso il Dipartimento di Neurologia Clinica, Istituto di Neurologia, Queens Square, Londra, Regno Unito, e Douglas C. Wallace presso la Emory University, Atlanta, GA.
La prima scoperta (Harding) ha identificato delezioni singole su larga scala del DNA mitocondriale (mtDNA) in biopsie muscolari di pazienti affetti da “miopatie mitocondriali” mentre la seconda scoperta (Wallace) ha identificato una mutazione puntiforme nel gene del mtDNA per la subunità 4 del complesso della NADH deidrogenasi (MTND4), associata alla neuropatia ottica maternamente ereditaria di Leber (LHON) in un ampio pedigree americano.
Queste due scoperte lanciarono una nuova fame di disturbi umani associati a mutazioni nella minuscola molecola del mtDNA: nello stesso anno, 1988, un folto gruppo di scienziati “mitocondriaci” alla Columbia University, guidato da Bud Rowland, chiarì l’eziologia genetica della sindrome di Kearns-Sayre (KSS), eponimo della scoperta di Anita Harding.
Bibliografia:
- “INTRAMITOCHONDRIAL FIBERS WITH DNA CHARACTERISTICS“, by Margit M. K. Nass and Sylvan Nass [J Cell Biol. 1963 Dec 1; 19(3): 593–611.]
- “Deoxyribonucleic acid associated with yeast mitochondria“, by G. Schatz, E. Haslbrunner, H. Tuppy [Biochemical and Biophysical Research Communications
Volume 15, Issue 2, 9 March 1964, Pages 127-132] - Wallace DC (1992) “Diseases of the mitochondrial DNA“ Ann. Rev. Biochem 61 : 1175 – 1212
- “Maternal inheritance of human mitochondrial DNA” by RICHARD E. GILES, HUGUES BLANC, HOWARD M. CANN, AND DOUGLAS C. WALLACE [Proc. Nati. Acad. Sci. USA
Vol. 77, No. 11, pp. 6715-6719, November 1980] - “Progress in understanding 2-hydroxyglutaric acidurias“, by Martijn Kranendijk, Eduard A. Struys, Gajja S. Salomons, Marjo S. Van der Knaap, and Cornelis Jakobscorresponding [J Inherit Metab Dis. 2012; 35(4): 571–587]
- El Hattab A.W & Fernando Scaglia (2013) “Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options”, Neurotherapeutics 10: 186 – 198
- “A Brief History of Mitochondrial Pathologies“, by Salvatore DiMauro [Int. J. Mol. Sci. 2019, 20(22), 5643]