Breve storia degli antibiotici chinolonici
 Sebbene non sia formalmente un chinolone, l’acido nalidixico è considerato il primo farmaco chinolonico. L’acido nalidissico è stato introdotto nel Regno Unito nel 1964, per il trattamento delle Infezioni delle Vie Urinarie (IVU) negli esseri umani. In senso tecnico, l’acido nalidissico è un naftiridone, non un chinolone: la sua struttura ad anello è un nucleo 1,8-naftiridinico che contiene due atomi di azoto, a differenza della chinolina, che ha un solo atomo di azoto.
Sebbene non sia formalmente un chinolone, l’acido nalidixico è considerato il primo farmaco chinolonico. L’acido nalidissico è stato introdotto nel Regno Unito nel 1964, per il trattamento delle Infezioni delle Vie Urinarie (IVU) negli esseri umani. In senso tecnico, l’acido nalidissico è un naftiridone, non un chinolone: la sua struttura ad anello è un nucleo 1,8-naftiridinico che contiene due atomi di azoto, a differenza della chinolina, che ha un solo atomo di azoto.
L’acido nalidixico è stato scoperto dal chimico statunitense George Y. Lesher (22febbraio 1926 – 17marzo 1990) e colleghi, che lavoravano presso la compagnia farmaceutica Sterling Drug Inc. (ora parte of Sanofi), in un distillato durante un tentativo di sintesi della clorochina, un farmaco utilizzato per la prevenzione e nel trattamento della malaria.
In una nota dell’articolo seminale del 1962 di Lesher e colleghi si affermava:
“In vitro ed in vivo fu scoperta attività antibatterica in una serie di derivati dell’acido 1-alchil-4-chinolone-3-carbossilico“.
ACIDO NALIDIXICO (O NALIDISSICO)
 La storia precisa della scoperta del primo chinolone dall’impurità originale della clorochina (che aveva una struttura chinolonica) all’acido nalidixico rimane avvolta nel mistero. Nella ricerca di un sottoprodotto per la sintesi della clorochina, Lesher scoprì dunque una molecola con modeste proprietà antibatteriche che sarebbe diventato il capostipite di un’importante famiglia di antibiotici. Ma Lesher non reclamò la scoperta dei chinoloni. Ciò sembra essere dovuto al fatto che composti chinolonici erano già stati brevettati da altri chimici ed altre aziende (ICI, Imperial Chemical Industries, ora AstraZeneca) verso la fine degli anni ’50, e prima ancora individuati da ricercatori australiani nel 1949.
La storia precisa della scoperta del primo chinolone dall’impurità originale della clorochina (che aveva una struttura chinolonica) all’acido nalidixico rimane avvolta nel mistero. Nella ricerca di un sottoprodotto per la sintesi della clorochina, Lesher scoprì dunque una molecola con modeste proprietà antibatteriche che sarebbe diventato il capostipite di un’importante famiglia di antibiotici. Ma Lesher non reclamò la scoperta dei chinoloni. Ciò sembra essere dovuto al fatto che composti chinolonici erano già stati brevettati da altri chimici ed altre aziende (ICI, Imperial Chemical Industries, ora AstraZeneca) verso la fine degli anni ’50, e prima ancora individuati da ricercatori australiani nel 1949.
Il primo brevetto di naftiridone della Sterling fu depositato gennaio 1961, nove mesi dopo la pubblicazione del brevetto di un chinolone fondamentale dell’ICI (“GB830832”) e quasi 4 anni dopo che ICI aveva presentato questo brevetto.
ACIDO NALIDIXICO e GB830832

Tuttavia, si è ragionevolmente portati a presumere che, a causa dei vincoli di proprietà intellettuale imposti dal precedente brevetto della ICI, la Sterling sia stata costretta a presentare una struttura di base non chinolonica (anche se strettamente correlata).
E’ anche vero che in questa storia di richiesta di primogenitura della scoperta dei chinolonici, un articolo tra i più citati è stato scritto da uno scienziato dell’AstraZeneca, azienda che non può affermare un ruolo terzo in tutta la vicenda.
L’acido nalidixico è quindi considerato il predecessore di tutti i membri della famiglia dei chinoloni, compresa la seconda, terza e quarta generazione comunemente noti come fluorochinoloni.
utilità clinica dell’acido nalidixico

Tuttavia, l’utilità clinica dell’acido nalidixico, nel trattamento di infezioni diverse dalle IVU, era limitata dalle sue basse concentrazioni sieriche e dalle elevate Concentrazioni Minime inibitorie (MIC).
Questo farmaco venne sostituito da molecole di seconda generazione con modifiche minori, inclusi composti come acido ossolinico, cinoxacina, acido pipemidico e altri introdotti negli anni settanta.
Tuttavia, nessuno di questi composti costituiva un importante progresso in termini di miglioramento dello spettro antibatterico o della farmacocinetica, e quindi i chinolonici rimanevano ancora limitati in gran parte al trattamento delle infezioni del tratto urinario.
Generazione di Chinoloni

Lo spettro d’azione dei chinoloni di prima generazione è limitato ad alcuni batteri Gram-negativi aerobi. I chinoloni di seconda generazione sono attivi nei confronti dei batteri Gram-negativi, anche resistenti, compresa la Pseudomonas aeruginosa.
L’aggiunta di una molecola di fluoro in C6 ha creato i “fluorochinoloni“. Questa categoria di agenti è uno dei gruppi di composti più intensamente studiati nel campo della medicina; infatti, dal rapporto originale di Lesher, è stato stimato che siano stati sintetizzati oltre 10000 analoghi dell’acido nalidixico o dei fluorochinoloni.
Nel 1998, Laura J. V. Piddock e colleghi della “University of Birmingham” suggeriranno che questi agenti (chinoloni e fluorochinoloni) hanno attività simili per gli enzimi topoisomerasi IV e DNA girasi, e che solo una mutazione in entrambi i geni conferirebbe un alto livello di resistenza a questi antibiotici.
i fluorochinoloni

Nel 1979 la pubblicazione di un brevetto depositato dal braccio farmaceutico della compagnia giapponese Kyorin Seiyaku Kabushiki Kaisha rivelò la scoperta della norfloxacina (AM-715) e la dimostrazione che alcune modifiche strutturali tra cui l’attaccamento di un atomo di fluoro all’anello chinolonico porta a una potenza antibatterica notevolmente migliorata.
Nonostante il sostanziale aumento dell’attività antibatterica della norfloxacina rispetto ai primi fluorochinoloni, non è diventato un antibiotico ampiamente utilizzato. Altre società hanno avviato programmi di scoperta di fluorochinoloni all’indomani della pubblicazione del brevetto di norfloxacina.
i fluorochinoloni (2)
La Bayer Pharmaceuticals ha scoperto che l’aggiunta di un singolo atomo di carbonio alla struttura della norfloxacina era in grado di fornire un altro miglioramento dell’attività, da 4 a 10 volte: la ciprofloxacina, brevettata nel 1980 è stata introdotta sul mercato farmaceutico nel 1987.
Un altro potente fluorochinolone, la levofloxacina, fu brevettata nel 1985 ed introdotta nel mercato farmaceutico giapponese nel 1993 ed in quello statunitense nel 1996. La levofloxacina è un fluorochinolone di terza generazione (che sono attivi contro le infezioni da Streptococcus pneumoniae e altri batteri Gram-positivi), essendo uno degli isomeri dell’ofloxacina, che era un analogo bloccato conformazionalmente a spettro più ampio della norfloxacina; sia l’ofloxacina che la levofloxacina sono state sintetizzate e sviluppate dagli scienziati della compagnia farmaceutica giapponese Daiichi Seiyaku.
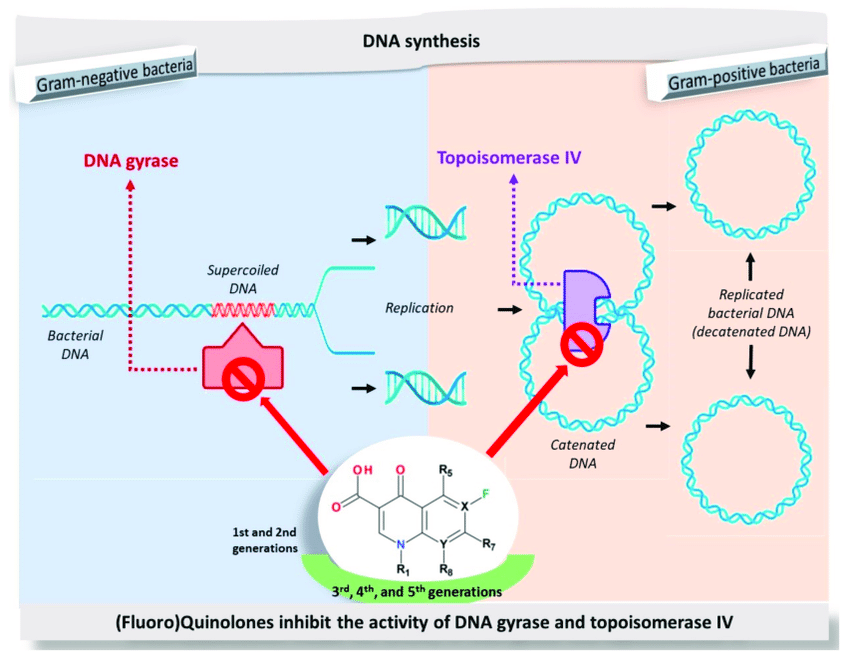
meccanismo d’azione sui batteri

I chinoloni inibiscono rapidamente la sintesi del DNA promuovendo la scissione del DNA batterico nei complessi DNA-enzima della DNA girasi e della topoisomerasi di tipo IV, con conseguente rapida morte batterica. L’organizzazione molecolare del complesso è attualmente sconosciuta sebbene siano stati suggeriti diversi modelli.
Sulla base di un modello biochimico, l’affinità dei chinoloni con gli ioni metallici sembra essere un importante prerequisito della loro attività antibatterica: probabilmente, i chinoloni si legano al complesso enzima-DNA tramite uno ione di magnesio.
FONTI E RIFERIMENTI:
- https://en.wikipedia.org/wiki/Quinolone_antibiotic#History
- Tweet del dott. Avraham Z. Cooper
- “1,8-NAPHTHYRIDINE DERIVATIVES. A NEW CLASS OF CHEMOTHERAPEUTIC AGENTS“; by G Y LESHER, E J FROELICH, M D GRUETT, J H BAILEY, R P BRUNDAGE (J. Med. Chem. 1962, 5, 5, 1063–1065 – Publication Date: September 1, 1962)
- “Origins of the Quinolone Class of Antibacterials: An Expanded “Discovery Story”” – Miniperspective – Gregory S. Bisacchi* (AstraZeneca, 35 Gatehouse Drive, Waltham, Massachusetts 02451, United States)
- “The Mysterious Origins of the Quinolones“, 16 MAR 2015 BY DEREK LOWE
- https://en.wikipedia.org/wiki/Nalidixic_acid
- https://www.newspapers.com/clip/40847304/obituary-for-george-yohe-lesher/
- “Quinolone antibiotics“, di Thu D. M. Pham,a Zyta M. Ziora b and Mark A. T. Blaskovich (Med. Chem. Commun., 2019, 10, 1719)
- “Quinolones: structure-activity relationships and future predictions“, di G. S. Tillotson (Pharmaceutical Division, Bayer plc, Strawberry Hill, Newbury, Berkshire RG13 1JA) pubblicato sul Journal of Medical Microbiology (01 May 1996 https://doi.org/10.1099/00222615-44-5-320)
- Norris S, Mandell GL (1988). “The quinolones: history and overview“. San Diego: Academic Press Inc. pp. 1–22.
- “Quinolones: Recent Structural and Clinical Developments“, di Saeed Emamia, Abbas Shafieeb and Alireza Foroumadib (pubblicato su Iranian Journal of Pharmaceutical Research (2005) 3: 123-136)
- “Chinoloni“, di Ilaria Randi (Ultima modifica 07.10.2019)
- “Activities of New Fluoroquinolones against Fluoroquinolone-Resistant Pathogens of the Lower Respiratory Tract“, di Laura J. V. Piddock,* M. Johnson, V. Ricci, and S. L. Hill (Antimicrob Agents Chemother. 1998 Nov; 42(11): 2956–2960.
doi: 10.1128/aac.42.11.2956) - https://www.kyorin-pharm.co.jp/en/company/history.shtml
- https://en.wikipedia.org/wiki/Norfloxacin
- “Comparative Activities of AM-715 and Pipemidic and Nalidixic Acids Against Experimentally Induced Systemic and Urinary Tract Infections“; Authors: Keiji Hirai, Akira Ito, Yashuo Abe, Seigo Suzue, Tsutomu Irikura, Matsuhisa Inoue, Susumu Mitsuhashi
- https://it.wikipedia.org/wiki/Ciprofloxacina
- https://en.wikipedia.org/wiki/Levofloxacin