LA STORIA DELLA GENOMICA: COME NASCE, COME SI SVILUPPA E COSA CI RISERVA IN FUTURO
di Sergio Barocci – Università di Genova per la terza età e divulgatore scientifico.
Il Sequenziamento del DNA (parte III)

E’ dalle prime iniziative del sequenziamento di interi genomi che nasce la genomica. Una probabile data di nascita la si può far coincidere nel 1980 con il sequenziamento del primo genoma: si trattava di un batteriofago.
Oggi, il sequenziamento degli acidi nucleici è diventato parte integrante della moderna ricerca biomedica. I progressi nella tecnologia di sequenziamento, dalla sua invenzione agli equivalenti moderni, sono stati straordinari.
Il 2022 segna, infatti, il 50° anniversario del sequenziamento del primo gene completo. È impressionante vedere fino a che punto è arrivata la scienza, ma proseguiamo per tappe.
Nel 1965, Holley sequenziò il primo tRNA (per l’alanina), per il quale venne insignito del Premio Nobel nel 1986. Egli determinò la struttura del tRNA utilizzando due ribonucleasi per dividere la molecola di tRNA in pezzi.
decifrazione del codice genetico

{kind=link}
Ogni enzima divideva la molecola nei punti di posizione per specifici nucleotidi la cui struttura venne manualmente “sconcertata” a mano, da un’intera squadra.
Dopo la decifrazione del codice genetico, si passò poi a una fase successiva, nella quale vennero messe a punto le prime tecniche utili per le colture cellulari che consentirono l’avvio dello sviluppo delle biotecnologie a partire dalla fine degli anni’60, con la scoperta da parte del microbiologo statunitense Hamilton O. Smith (23agosto 1931 – ) degli enzimi di restrizione (riconoscono e scindono il DNA in corrispondenza di specifiche sequenze palindromiche, ossia sequenze simmetriche di 4 – 6 bp (base pair), lette sulle due emi-eliche di DNA in direzione 5’ – 3’ e viceversa) con i quali diventò possibile tagliare, isolare e introdurre un particolare gene nel DNA di un microrganismo, ottenendo il primo DNA ricombinante nel 1972.
DNA RICOMBINANTE

E’ agli inizi degli anni ’70 che nacque infatti la tecnologia del DNA ricombinante, grazie al lavoro del biochimico statunitense Paul Berg (1926 – ) che gli valse il Premio Nobel nel 1980 per aver prodotto nel 1972 la prima molecola di DNA ricombinante utilizzando enzimi di restrizione e in seguito nel 1973 grazie ai lavori del biotecnologo e imprenditore statunitense Herbert W. Boyer ( 1936 – ) e del genetista statunitense Stanley Cohen (1935 – ) che dimostrarono che gli enzimi di restrizione potevano essere utilizzati come “forbici ” per tagliare frammenti di DNA, per poi essere inseriti in un vettore-plasmide. Essi, infatti, clonano il primo gene animale usando per la prima volta un plasmide (i plasmidi sono molecole di DNA circolare a doppia elica che si trovano di norma in alcuni batteri; sono di dimensioni variabili da due a parecchie centinaia di kilobasi o Kb e portano geni per l’inattivazione di antibiotici, per la produzione di tossine e per la degradazione di prodotti naturali).
ENZIMI DI RESTRINZIONE

Nel 1973, Boyer e Cohen dimostrarono che gli enzimi di restrizione avrebbero potuto essere utilizzati come “forbici” per tagliare frammenti di DNA di interesse da una fonte, per poi essere inseriti in un vettore – plasmide.
Mentre Cohen proseguì i suoi studi ritornando nell’ambito del mondo accademico, Robert A. Swanson (1947–1999) venne contattato da Boyer per fondare la Genentech Inc.
Boyer lavorò anche con il genetista statunitense Arthur Riggs (8agosto 1939 – 23marzo 2022) ed il chimico giapponese Keiichi Itakura (18febbraio 1942 – ) della Beckman Research Institute, diventando i primi scienziati ad esprimere con successo un gene umano nei batteri producendo l’ormone somatostatina nel 1977.
Al gruppo si aggiunsero, in seguito, il biologo statunitense David Goeddel (1951 – ), il biochimico italo- statunitense Roberto Crea (1luglio 1948 – ) e il chimico statunitense Dennis Kleid che contribuirono alla produzione di insulina umana sintetica nel 1978.
sequenze furono determinate a livello di RNA

Un ulteriore passo avanti fu fatto quando, nello stesso anno, il biologo belga Walter Fiers (31gennaio 1931 – 28luglio 2019) nel 1972 divenne il primo a sequenziare il gene che codifica per la proteina del mantello del batteriofago MS2 le cui sequenze furono determinate a livello di RNA.
Ma la ricerca più importante per lo sviluppo del sequenziamento del DNA venne fatto dal chimico britannico Fredrick Sanger (1918 – 2013) nel 1977, quando pubblicò un nuovo ed efficiente metodo detto terminazione a catena “dideoxy” per sequenziare il DNA mediante interruzione controllata della sua replicazione, noto anche come “Sanger Sequencing”.
ll primo genoma completo ad essere sequenziato manualmente senza alcuna automazione da F. Sanger fu il batteriofago phiX174 di circa 5000 basi, che aprì la porta al campo della genomica.
sequenziamenti di genomi

Questa impresa fu di notevole importanza in quanto portò alla creazione, nel 1982, del Progetto “Gen Bank“, un database open access, istituito da una sezione del National Institute of Health (N.I.H) e dal National Center for Biotechnology Information (NCBI) allo scopo di raccogliere e mettere a disposizione di tutti le sequenze di nucleotidi e le relative proteine ottenute dopo la loro traduzione.
Dopo questo successo, Sanger nel 1984 decifrò anche la sequenza completa del DNA del virus EBV, scoprendo che conteneva 172.282 nucleotidi.
Sempre nel 1977, i biochimici statunitensi Allan Maxam e Walter Gilbert (21marzo 1932 – ) svilupparono un altro metodo di sequenziamento del DNA detto “chimico“. Questo metodo pur diventando subito popolare e molto impiegato venne, a poco a poco, accantonato a causa del successivo miglioramento del metodo di Sanger che non impiegava composti chimici dannosi alla salute, rivelandosi sin da subito più efficiente con una lettura fino a 800 basi contemporaneamente.
Conoscenza delle sequenze di DNA

Il metodo dei terminatori a catena o dell’interruzione controllata della replicazione, valse a Sanger il secondo Premio Nobel per la chimica nel 1980, premio condiviso con Gilbert e con Berg, dopo quello ottenuto nel 1958 per aver sequenziato la struttura aminoacidica dell’insulina.
La conoscenza delle sequenze di DNA incominciò così a diventare indispensabile per la ricerca biologica di base ma anche in numerosi campi applicati come la diagnostica, le biotecnologie, la biologia forense, e la biologia sistematica.
Il metodo di Sanger, nonostante si dimostrasse efficiente, era, però, ancora manuale, molto lento ed estremamente laborioso, soggetto spesso anche all’errore umano e legato all’uso di sostanze radioattive dannose per la salute. Quindi, era indispensabile apportare dei miglioramenti, anche nell’eventualità di sequenziare genomi di organismi molto più grandi di un virus, come i protozoi, gli animali e gli umani.
elettroforesi capillare e pcr

Il superamento di queste problematiche avvenne, dopo alcuni anni, con l’uso della rivoluzionaria tecnica dell’elettroforesi capillare che sostituì la precedente tecnica elettroforetica in gel di poliacrilamide, dove i frammenti separati erano marcati con fluorofori o fluorocromi (molecole che dopo aver assorbito fotoni o quanti di energia elettromagnetica di una certa lunghezza d’onda emettono fluorescenza) al posto di elementi radioattivi .
Questa permise un notevole sviluppo tecnologico che, supportato dall’uso della tecnologia PCR (Polymerase Chain Reaction) inventata dal biochimico statunitense Kary Mullis (1944 – 2019) nel 1983, consentì l’implementazione di sequenziatori automatici che impiegarono questi sistemi di rilevazione della fluorescenza incrementando notevolmente le capacità di analisi, segnando l’inizio della ricerca genomica.
introduzione di varianti

Nel 1986, il laboratorio del biologo statunitense Leroy Hood (1938 – ), presso il California Institute of Technology in collaborazione con Applied Biosystems (ABI), pubblicava il primo rapporto sul sequenziamento automatizzato del DNA che introduceva una variante, cioè una colorazione fluorescente al posto della marcatura radioattiva per ciascuno dei quattro terminatori a catena dideossinucleotidi permettendo il sequenziamento in una singola reazione anziché in quattro. In aggiunta, questo iniziale rapporto mostrava che i risultati del sequenziamento potevano essere raccolti direttamente su un computer.
Il sequenziamento di Sanger con i coloranti fluorescenti divenne la tecnica di sequenziamento dominante sino all’introduzione delle tecnologia di nuova generazione (NGS) a partire dal 2005. La lunghezza della sequenza ottenibile era ora di circa 1000 nucleotidi.
la prima macchina di sequenziamento completamente automatizzata

Nel 1987 venne annunciata la commercializzazione, da parte della ditta Applied Biosystems, della prima macchina di sequenziamento completamente automatizzata, l’ABI 310. Questa macchina utilizzava un processo di ricarica automatica di capillari con una matrice polimerica, in sostituzione del lungo e dispendioso lavoro di versare il gel nelle piastre come avveniva in precedenza.
I frammenti di DNA marcati correvano lungo il capillare, con differenti velocità, fino a raggiungere una zona detta “finestra del capillare” con maggiore trasparenza, dove un raggio laser eccitava i quattro fluorocromi con emissione di lunghezze d’onda diverse che venivano rilevate da una cella fotoelettrica in sequenze genomiche colorate con diverse intensità luminose e registrate in forma grafica (cromatogramma).
l’ABI

In breve, il risultato corrispondeva ad una sequenza di picchi di quattro colori diversi, uno per ogni nucleotide. L’ABI 310 Genetic Analyzer possedeva un unico capillare a cui ben presto seguirono macchine a 4, 16 e infine a 48 o 96 capillari come l’ABI 373 che permise il sequenziamento di interi genomi. Con questi dispositivi automatizzati si potevano sequenziare frammenti di circa 500.000 bp al giorno, con una lunghezza massima per frammento di circa 600 basi e riducevano significativamente le possibilità di errori.
I progressi nel sequenziamento del DNA vennero soprattutto aiutati anche dallo sviluppo concomitante della tecnologia del “DNA ricombinante“ che permise ai campioni di DNA di poter essere isolati da fonti diverse dai virus. La messa in commercio di kit per il sequenziamento automatico pronti all’uso, rese per molto tempo il metodo di Sanger il “gold standard” per il sequenziamento. Questa procedura, oltre ad un risparmio in termini di tempo, permise l’analisi simultanea di più campioni.
LO SHOTGUN

Nel corso degli anni ’80 si mise anche a punto un metodo di clonazione non selettivo chiamato “shotgun” ; con esso si potevano produrre vari frammenti di DNA a caso che venivano clonati per la produzione di librerie in Escheria coli. I cloni erano poi sequenziati casualmente e i risultati assemblati per ottenere la sequenza genomica, poi completata da un software che confrontava tutte le lettere della sequenza e allineava le sequenze corrispondenti.
Lo shotgun diventò , in quel periodo, il metodo di elezione per l’analisi del genoma. Tale metodo, venne successivamente esteso con l’utilizzazione di vettori di clonazione BAC “Bacterial Artificial Chromosome“ (vettori artificiali di DNA basati sul plasmide F isolato da Escherichia coli), YAC “Yeast Artificial Chromosome” (cromosomi artificiali di lievito utilizzati nella tecnologia del DNA ricombinante) in cui si potevano inserire frammenti di DNA sempre più grandi.
Nel 1991, il biologo statunitense Craig Venter (1946 – ) diede inizio al sequenziamento di sequenze di cDNA chiamate ES “Expressed Sequence Tag” utilizzate per identificare i trascritti genici nel tentativo di catturare la frazione codificante del genoma umano. Queste due ultime innovazioni insieme allo shotgun sequencing , fornirono una spinta fondamentale per l’avvio del Progetto Genoma iniziato nel 1990 e terminato nel 2003.
IL PROGETTO GENOMA UMANO

Il Progetto Genoma o HGP “ Human Genome Project” fu realizzato dall’U.S. Department of Energy (DOE) e dal National Institute of Health (NIH) con la partecipazione di strutture di Tecnologia e Ricerca di tutto il mondo. Ebbe una durata di 13 anni, dal 1990 al 2003 e come obiettivi la determinazione della sequenza dell’intero genoma umano, la definizione di tutti i geni in esso presenti, il corretto ordine di tutti i tre miliardi di basi del genoma umano e la creazione di un database per raccogliere tutte queste informazioni da mettere a disposizione di Enti di ricerca pubblici e privati per studi successivi. Il progetto è stato completato nel 2003 confermando che gli esseri umani hanno 20.000-25.000 geni.
LA CELERA GENOMICS

Inizialmente si trattò di un progetto pubblico guidato da due istituzioni statunitensi, ma, quando nel 1998 Venter lasciò il National Institute of Health (NIH) per fondare la Celera Genomics (Società di ricerca privata), egli proseguì, per fini commerciali, il progetto di caratterizzazione del genoma umano. Lo scopo di tale società privata, infatti, era quello di creare una banca dati genomica che poteva essere utilizzata a seguito del pagamento di una determinata tariffa.
Tale approccio rese Venter molto impopolare tra la comunità scientifica e, soprattutto, ebbe l’effetto di dare ulteriore vigore ai numerosi gruppi che stavano partecipando al Progetto pubblico coordinato dal genetista statunitense Francis Collins ( 1950 – ) dell’NIH (National Institute of Health).
Per portare a termine il sequenziamento del genoma umano, i laboratori della Celera Genomics misero a punto, nel 1999, la tecnica dello “shotgun sequencing”, che permise a Venter di annunciare nel 2000, in presenza del Presidente degli Stati Uniti d’America, Bill Clinton, il quasi completamento del lavoro in concomitanza con il contemporaneo annuncio di Collins, direttore del Progetto Genoma Umano.
whole genome shotgun
La metodica dello shotgun sequencing si fondava sul metodo enzimatico di Sanger tramite i nuovi sequenziatori automatici dell’Applied Biosystems che permettevano di codificare 400.000 basi al giorno, riducendo notevolmente i tempi necessari all’ottenimento di una sequenza. La Celera Genomics elaborò ulteriormente un approccio alternativo di sequenziamento, definito whole genome shotgun, in cui il genoma veniva spezzettato in numerosi frammenti casuali di diverse dimensioni (da 5 a 20 Kb e da 0,4 a 1,2 Kb) chiamate “ reads”. La procedura veniva ripetuta più volte allo scopo di avere delle reads parzialmente sovrapposte e la sequenza risultante era ottenuta assemblando, al calcolatore, le diverse reads.
Le strategie di sequenziamento durante il Progetto Genoma Umano
Durante la fase finale del progetto e negli anni successivi, furono avviati diversi studi tra cui il Progetto Internazionale HapMap per sviluppare una mappa aplotipica del genoma umano ed il 1000 Genomes Project per una maggiore comprensione della variazione genetica negli esseri umani. A partire dal mese di settembre del 2003 si aggiunse un altro progetto internazionale denominato “Encode” (Encyclopedia of DNA elements) con l’obiettivo di trovare e catalogare tutti gli elementi funzionali presenti nel genoma umano di organismi modello, in maniera estesa ed enciclopedica.
L’avvento dell’automazione

L’avvento dell’automazione, alla fine degli anni ’80, rese il sequenziamento di interi genomi molto più veloce e contribuì alla commercializzazione di sequenziatori automatici che, ben presto si diffusero in tutta la comunità di ricerca scientifica. Il primo sequenziatore automatico commerciale, a elettroforesi capillare, venne introdotto nel 1997 dall’Applied Biosystems, l’ABI 310.
Quando fu lanciato il Progetto Genoma Umano negli anni ’90, si incominciò ad avviare su larga scala lo studio sul sequenziamento dei primi genomi batterici e arcaici. A quel tempo esistevano due approcci per sequenziare un genoma. Il primo (“hierarchical shotgun”) creava inizialmente una mappa che permetteva di suddividere il genoma in diversi segmenti attraverso la costituzione di mappe genetiche e fisiche e, successivamente, proseguiva con il sequenziamento dei vari segmenti.
hierarchical shotgun

Si può trovare un’analogia a questo approccio pensando di ordinare i capitoli di un libro e poi trovare le parole comprese in ogni capitolo. Tale tecnica fu, per esempio, quella utilizzato dal Consorzio Pubblico del Progetto Genoma Umano che si basava su mappe di cloni contigui all’interno di vettori artificiali batterici BAC o YAC.
Con questo metodo, il DNA umano veniva frammentato in pezzi (100.000 – 200.000 bp) e i frammenti poi clonati in batteri che conservavano e replicavano il DNA in modo da prepararne quantità abbastanza grandi per il sequenziamento. Ogni clone BAC veniva mappato in modo da determinare il punto del genoma umano da cui originava il DNA del clone stesso. Questo metodo assicurava la comprensione della sequenza precisa del DNA di ogni clone e la relazione spaziale con gli altri cloni. Minimizzando la sovrapposizione, occorrevano circa 20.000 cloni BAC diversi per raccogliere i 3 miliardi di bp del genoma umano.
Per il sequenziamento, ogni clone BAC veniva tagliato in frammenti più piccoli (circa 2000 basi) che , in seguito, erano sequenziati automaticamente da uno strumento che leggeva circa 500 -800 basi a reazione. Dopo che ogni frammento era stato sottoposto a sequenziamento, un computer allineava le sequenze contigue permettendo in tal modo di risalire alla sequenza di un clone BAC.
sequenziamento diretto

Il secondo approccio, denominato “WGS” (Whole-Genome Shotgun), ovvero sequenziamento diretto, prevedeva la frammentazione casuale del genoma in segmenti parzialmente sovrapposti e poi il loro sequenziamento. La sequenza genomica era in seguito assemblata da un computer sulla base delle sovrapposizioni di sequenza tra i frammenti. Questo approccio era analogo a quello di strappare diverse copie dello stesso libro per poi ricostruirne una copia completa rimettendo insieme le pagine che si sovrapponevano.
Con tale modalità, nel 1995 Venter, in collaborazione con Smith, sequenziò il genoma completo (1,8 Mb) di Haemophilus influenzae, (un batterio Gram negativo a forma di bastoncino immobile descritto per la prima volta nel 1892 in seguito ad una pandemia di influenza), senza far uso di mappe fisiche e genetiche e di Mycoplasma genitalium con genoma di 580 Kb. Il successivo passo fu il sequenziamento, nel 1997, del genoma del batterio Bacillus subtilis di 4,2 Mb. L’ introduzione dello shotgun sequencing permise di estendere lo studio ad altri genomi. Tra questi, nel 1996, vi fu anche quello del primo eucariota unicellulare, il lievito di birra “Saccharomyces cerevisiae“ di 12 Mb.
pirosequenziamento ed high-throughput
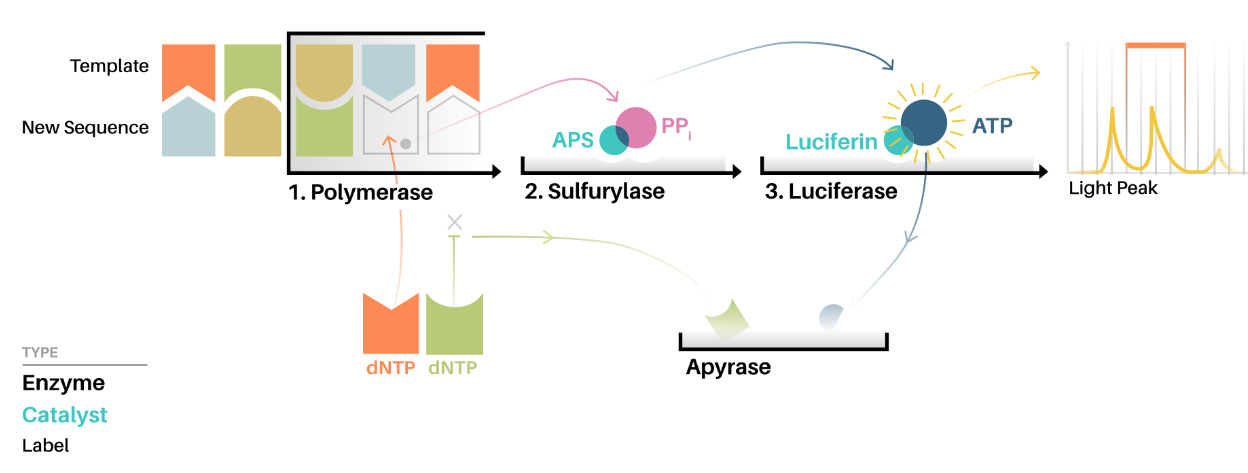
Nello stesso anno il biochimico svedese Pal Nyren (1955 – ) e il biologo molecolare iraniano Mostafa Ronaghi (1968 – ), al Royal Institute of Technology di Stoccolma, pubblicavano il metodo del pirosequenziamento.
A partire da quell’anno furono sviluppati i metodi per il sequenziamento “high –throughput“. Infatti, nel 1997, il biofisico francese Pascal Meyer e il biologo svizzero Laurent Farinelli presentarono un loro brevetto alla Word Intellectual Property Organization implementato successivamente nei sequenziatori Hi Seq Illumina mentre i genetisti molecolari statunitensi Frederick Blattner e Guy Plunkett dell’Università del Wisconsin completavano la sequenza dell’Escherichia Coli di 5 Mb.
Nel 2000, la Lynx Therapeutics (acquistata nel 2004 dalla Solexa Inc) pubblicava e lanciava sul mercato il sequenziamento “MPSS” (Massively Parallel Signature Sequencing); un processo tecnologico avanzato di sequenziamento massivo parallelo utilizzato per identificare e quantificare i trascritti degli mRNA, dando inizio al sequenziamento di nuova generazione o “ NGS “.
Massively Parallel Signature Sequencing

Durante il 1998 venne sequenziato, da parte del biologo britannico John Sulston (1942 – 2018) (Premio Nobel per la Medicina nel 2002) del Sanger Center in collaborazione con l’Università di Washington, il genoma del nematode Caenorhabditis elegans (100 Mb) , primo esempio di sequenziamento di un organismo eucariotico multicellulare.
Inoltre, sempre in quell’anno, fu sequenziata una prima bozza della mappa del genoma umano che mostrava la bellezza di 30.000 geni. Il biologo computazionale statunitense P. Green dell’Università di Washington (Seattle), pubblicava un programma informatico denominato “Phred” sviluppato per verificare la bontà del sequenziamento (analisi degli elettroferogrammi). Phred ha svolto un ruolo particolarmente notevole anche nel Progetto Genoma Umano in cui grandi quantità di dati di sequenza erano elaborati da script automatizzati, insieme ad un altro software “Phrap”, un programma per assemblare le sequenze di DNA.
phred e phrap

Nel 1999 fu sequenziato il cromosoma umano 22 e nel 2000, la sequenza completa del cromosoma 21. In successione, quella del genoma del moscerino della frutta Drosophila Melanogaster, del genoma del pesce Fugu rubripes e il primo genoma vegetale, quello dell’organismo modello Arabidopsis thaliana (125 Mb ).
Nel febbraio del 2001, su Science veniva pubblicata la bozza del progetto genoma umano da parte di Celera seguita dopo nel mese (giugno) dalla pubblicazione, su Nature, quella del Consorzio Pubblico. Il 2002 fu l’anno in cui venne invece annunciato il sequenziamento completo del genoma murino del topo da laboratorio Mus musculus mentre nel giugno del 2003 c’è stato l’annuncio ufficiale del completamento del Progetto da parte dell’UCSC Genome Bioinformatics Group con due anni di anticipo rispetto ai tempi stabiliti. Nel maggio del 2006, si è aggiunta un’altra pietra miliare, quando la sequenza dell’ultimo cromosoma umano, il cromosoma 1 (il più lungo e il più difficile da analizzare) è stata resa pubblica via Internet dall’NIH.
sequenziamenti di cromosomi umani

Le principali scoperte del Progetto Genoma Umano sono risultate le seguenti: a) Il genoma umano è lungo circa 3200Mb, di cui solo 48Mb di DNA codificante (1,5%), 1152Mb (36%) costituiscono il cosiddetto “gene related” DNA (Introni, UTR, Pseudogeni, frammenti genici) e le restanti 2000Mb costituiscono il DNA intergenico, formato da sequenze ripetute intersperse chiamate LINE (21%), SINE (15%), LTR (9%), Trasposoni a DNA (3%), sequenze ripetute in tandem anche dette microsatelliti (3%) e una piccola percentuale di altri tipi; b) i geni umani sono circa 20-25000, in media contengono 8.8 esoni ognuno dei quali è lungo all’incirca 170bp. Il numero di introni è in media 7.8 con una lunghezza media di 5420bp.
In realtà, dal suo rilascio iniziale nel 2000, il genoma umano di riferimento ha coperto solo la frazione eucromatica del genoma, lasciando importanti regioni eterocromatiche incompiute, in quanto circa l’8% del totale circa 200 milioni di bp non erano state sequenziate.
filling the gaps

Solo nel 2022 è avvenuto il completamento da parte del gruppo del Consorzio Telomere- to- Telomere o T2T della parte non sequenziata in aggiunta alla correzione di alcuni errori precedenti. Una sequenza più completa e senza lacune del genoma umano è stata eseguita per mezzo di nuovi metodi di sequenziamento del DNA e di analisi computazionale e pubblicata con sei articoli e copertina dedicata dal titolo “ Filling the gaps” sulla Rivista Science del 31 Marzo – 1° aprile del 2022.
La versione completa del genoma umano è risultata composta da 3.055 miliardi di coppie di basi, le unità da cui sono costruiti i cromosomi e i nostri geni e 19.969 geni che codificano per le proteine. Di questi geni, ne sono stati identificati circa 2.000 nuovi. La maggior parte di questi risulta disabilitata ma 115 potrebbero essere ancora attivi. Sono state, inoltre, individuate circa 2 milioni di varianti genetiche aggiuntive, 622 delle quali erano presenti in geni clinicamente rilevanti.
Lo shotgun sequencing
Nella metodologia “shotgun sequencing”, detto anche a colpo di fucile, venivano prodotti frammenti di DNA a caso; era un metodo radicalmente nuovo e differente da quello “gerarchico” che era più indaginoso in quanto prevedeva la costruzione di una libreria BAC e di una mappa fisica. Lo shotgun sequencing si dimostrava più semplice da un punto di vista della preparazione della libreria genomica (più veloce e meno costoso) ma più esigente dal punto di vista computazionale; questo problema fu risolto solo con lo sviluppo di metodi bioinformatici più avanzati.
L’utilizzo del metodo shotgun per il sequenziamento di genomi di più grandi dimensioni pose diversi problemi di assemblaggio dei contigs (tratti di sequenza assemblati senza discontinuità) per le sequenze ripetute; per questo motivo, nei primi anni del 2000, cominciarono ad apparire tecnologie innovative capaci di effettuare un elevatissimo numero di sequenziamenti in contemporanea (anche dell’ordine di miliardi) e denominate “tecnologie ad elevato parallelismo”. Questa evoluzione ha determinato la nascita e lo sviluppo di molteplici tecnologie dotate di alta processività raggruppate sotto la denominazione di Next Generation Sequencing o NGS.
Next Generation Sequencing
Le tecniche NGS hanno promosso nuove tecnologie hight – throughtput applicate nel campo della genomica, metagenomica, trascrittomica e metatrascrittomica che, a differenza di quelle precedenti, consentirono di ottenere una descrizione più dettagliata degli ecosistemi microbici.
Esse, possono essere utilizzate sostanzialmente in due modi:
1. sequenziamento di tutto l’acido nucleico microbico
2. sequenziamento di uno specifico gene
Il primo ha fornito informazioni, oltre che della composizione filogenetica, anche del numero e della potenziale funzione di specifici geni, mentre il secondo ha consentito l’amplificazione di un gene specifico tramite PCR per isolare le regioni genomiche di interesse, in modo da ottenere una quantità di DNA necessaria per il sequenziamento.
clicca per andare alla quarta ed ultima parte
Bibliografia:
- Sanger F.& Coulson R., (1975). “A rapid method for determining sequences in Dna by primed synthesis with Dna polymerase” . Journal of molecular biology, 94, 441 – 448.
- Sanger F., Nicklen S., Coulson A. R., 1977. “DNA -sequencing with chain-terminating inhibitors.” PNAS 74, 5463 – 5467.
- Maxam A.M. & Gilbert W. (1977) “A new method for sequencing DNA” PNAS 74 : 560 – 564
- Mullis K.F., et al. (1986) “Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction“ , in Cold Spring Harbor Symposia on Quantitative Biology, vol. 51, pp. 263 – 273.
- Smith LM. et al. (1986). “Fluorescence detection in automated DNA sequence analysis“. Nature. 321 (6071): 674 – 679.
- The International HapMap Project (2003) ” Nature. 426 (6968): 789 – 796
- International HapMap Consortium (2005). “A haplotype map of the human genome“. Nature. 437 (7063): 1299 –1320.
- International HapMap Consortium (2007). “A second generation human haplotype map of over 3.1 million SNPs“. Nature. 449 (7164): 851 – 861.
- International HapMap 3 Consortium (2010). “Integrating common and rare genetic variation in diverse human populations“. Nature. 467 (7311): 52 – 58.
- Durbin, R. M. et al. (2010). “A map of human genome variation from population-scale sequencing“. Nature. 467 (7319): 1061–1073.
- McVean, G. A et al. (2012). “An integrated map of genetic variation from 1,092 human genomes” Nature. 491 (7422): 56 – 65
- Birney E. et al. (2007). “Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project” Nature. 447 (7146): 799 – 816.
- Fleischmann R.D. et al.(1995) “Whole-genome random sequencing and assembly of Haemophilus influenza” . Science 269: 496 – 512.
- Gofieau A. et al. (1996) “Life with 6000 Genes” Science 274, 546-567
- Blattner F.R., et al. (1997) “The Complete Genoma Sequence of Escherichia coli K-12“ Science 277, 1453 – 1462
- Hodgkin J.. et al. . (1998) “C. elegans: Sequence to Biology“ Science 282, 2011 – 2018
- Ewing B. et al. (1998). “Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment” Genome Res. 8 , 175 – 185
- Dunham I. . et al. (1999) . “The DNA sequence of human chromosome 22 .” Nature, vol. 402 , 489 – 495
- Hattori M… et al. (2000). “The DNA sequence of human chromosome 21.” Nature, vol. 405, 311 – 319.
- Adams M.D. .et al. (2000) “The genome sequence of Drosophila melanogaster”. Science. 287, 2185 – 2195.
- Aparicio S. et al (2002). “Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes”. Science 297, 1301 – 1310
- The Arabidopsis Genome Initiative (2000). ”Analysis of the genome sequence of the flowering plant Arabidopsis thaliana”. Nature 408, 796 – 815.
- Lander E.S. et al. (2001) “Initial sequencing and analysis of the human genome“ Nature 409(6822): 860-921
- Venter J.C. et al. (2001), ”The sequence of the human genome, in Science” 291, 1304 – 1351