I casi clinici del New England Journal of Medicine: Pachidermoperiostosi
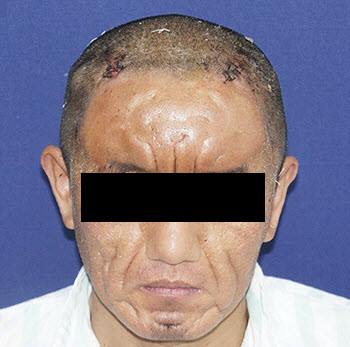 Caso clinico del NEJM: Un trentaduenne ha presentato un aspetto “particolare” del viso. Dopo la pubertà egli aveva notato un progressivo ingrandimento delle sue mani e piedi oltre a “corrugamento” facciale.
Caso clinico del NEJM: Un trentaduenne ha presentato un aspetto “particolare” del viso. Dopo la pubertà egli aveva notato un progressivo ingrandimento delle sue mani e piedi oltre a “corrugamento” facciale.
Il paziente ha riferito che la progressione della malattia si era stabilizzata dopo i 27 anni.
All’esame obiettivo egli presentava eccessiva secrezione sebacea, la pelle della fronte, delle guance e del mento rugosa in maniera ridondante, oltre ad “ingrandimento cilindrico” delle dita degli arti.
Il paziente ha ricevuto diagnosi clinica di pachidermoperiostosi, una rara sindrome genetica caratterizzata da ispessimento cutaneo, “clubbing digitale” e periostosi.
I parenti non hanno sintomi e segni simili e non è stato eseguito alcun test genetico.
La terapia da parte del personale medico dell’ospedale “Changzheng” di Shangai è stata eseguita in due fasi: nella prima fase è stato impiantato un “expander” sotto la cute della fronte del paziente per far espandere ed assicurare un adeguato tessuto cutaneo mentre la resezione della cute “rugosa e ridondante” è stata eseguita nella seconda fase.
UNA MALATTIA OSSEA EREDITARIA RARA
Altri nomi di questa rara sindrome genetica che interessa ossa e cute sono Osteopatia Primaria Idiopatica o Osteartropatia Ipertrofica Idiopatica o Sindrome di Touraine-Solente-Golè.
Nel 1868 la malattia venne descritta da Friedreich come “eccessiva crescita delle ossa dell’intero scheletro”. Touraine, Solente e Golè, tre dermatologi, descrissero nel 1935 la Pachidermoperiostosi come la forma primaria di una malattia ossea ipertrofica e la distinsero in tre forme (completa, incompleta e frusta).
Riferimenti:
- https://www.nejm.org/doi/pdf/10.1056/NEJMicm1309107
- https://en.wikipedia.org/wiki/Primary_hypertrophic_osteoathropathy
- “Pachydermoperiostosis: an update“, di M Castori, L Sinibaldi, R Mingarelli, RS Lachman, DL Rimoin, B Dallapiccola (First published: 18 October 2005 https://doi.org/10.1111/j.1399-0004.2005.00533.x)
- “Pachydermoperiostosis (Touraine–Solente–Gole syndrome): a case report“, di
Amir Joshi, Gaurav Nepal, Yow Ka Shing, Hari Prasad Panthi, and Suman Baral (J Med Case Rep. 2019; 13: 39. – Published online 2019 Feb 21. doi: 10.1186/s13256-018-1961-z)